Hydride Reduction Reactions: A Stereoselective Adventure
Written by Ryan
Introduction
The purpose of this experiment is to investigate the stereoselectivity of various reducing agents in the reduction of a carbonyl group. The first part of this experiment will focus on the stereoselectivity of sodium borohydride, lithium aluminum hydride, and lithium tri-sec-butylborohydride (L-selectride) in the reduction of 4-tert-butylcyclohexanone.1 The second part of this experiment will focus on the stereoselectivity of saccharomyces cerevisiae (baker’s yeast) in the reduction of ketone in ethyl acetoacetate.2 The stereoselectivity of each reducing agent will be examined via 1H-NMR spectroscopy, and the product will be further characterized using infrared (IR) spectroscopy as well as melting point analysis. Figure 1 below shows the structures of the metal hydride reducing agents.
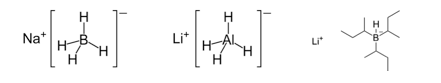
Figure 1: Structure of sodium borohydride, lithium aluminum hydride, and L-selectride, respectively.
Metal hydride reduction of a carbonyl group to an alcohol involves activation of the carbonyl carbon by the metal followed by the nucleophilic attack of hydride to the carbonyl carbon. The work up is commonly performed in H2O or H3O+ to protonate the oxygen atom and form the alcohol OH group.3 A reaction scheme for the reduction of 4-tert-butylcyclohexanone is shown below.
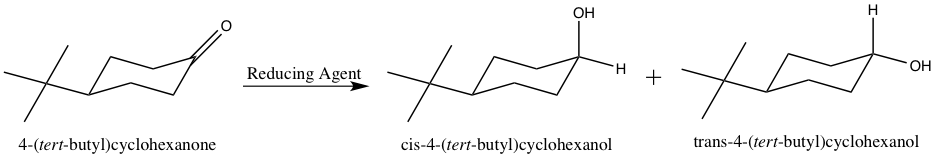
Figure 2: Reduction of 4-tert-butylcyclohexanone.
The cis and trans products in Figure 2 show the resulting alcohol OH group in the axial and equatorial positions, respectively. Due to the sp2 hybridization of the carbonyl carbon, it is planar, and thus, can be attacked from either the top or bottom face of the molecule. By attacking from the top face of the molecule, the resulting alcohol OH group is pushed to the equatorial position, as shown in the trans product above. On the contrary, attacking from the bottom face of the molecule pushes the alcohol OH group into the axial position, as shown in the cis product above. The trans product is the thermodynamically more stable product due to the tert-butyl and alcohol OH groups both being in the equatorial positions. The equatorial position results in less steric crowding, and thus, it is favorable for larger substituents to occupy that position.3 However, the cis product forms more rapidly due to the bulky tert-butyl group impeding the ability of the hydride ion to attack the top face of the molecule.
Being that the trans product is more thermodynamically stable, it is said to be under thermodynamic control. Thermodynamic control means the product that is more energetically stable will form preferential to other, less stable products.3 However, the formation of the thermodynamic product involves a greater activation energy.4 The cis product involves a smaller activation energy, and forms more rapidly than the thermodynamic product.4 Thus, the cis product is regarded as being under kinetic control. Kinetic control means the product that forms the most rapidly is favored, despite not being the most stable.3 The formation of a thermodynamically favored product is reversible, and the formation of a kinetically favored product is irreversible.4 By analyzing the 1H-NMR spectra of the hydride reduction products, the ratio of trans to cis product formed in each reduction reaction can be determined.
Understanding each metal hydride reducing agent allows for an educated guess as to whether the hydride will attack from the top or bottom face of the 4-tert-butylcyclohexanone. Both sodium borohydride and lithium aluminum hydride are smaller, less bulky metal hydride reducing agents, and thus it is expected that they will be able to deliver the hydride from the top face of the molecule (see “Figure 1” and “Figure 2”).4 The bulky tert-butyl group will not hinder the hydride attacking from the top face. However, L-selectride is a much larger and bulkier metal hydride reagent, and thus, would most likely not be able to attack from the top face with the presence of the bulky tert-butyl group (see “Figure 1” and “Figure 2”).4 Accordingly, the hypothesis is that the sodium borohydride and lithium aluminum hydride will form the trans product, and the L-selectride will form the cis product.
Baker’s yeast contains naturally occurring biochemical enzymes with reductive capabilities. This experiment focuses on the yeast reduction of ethyl acetoacetate.2 The reaction scheme for the yeast reduction of ethyl acetoacetate is shown below.
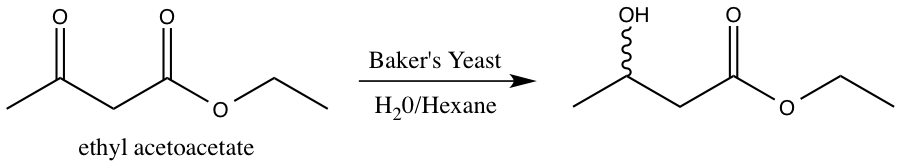
Figure 3: Reduction of ethyl acetoacetate using baker’s yeast.
The enzymes present in baker’s yeast selectively reduce the ketone over the ester. The ester is more difficult to reduce due to the presence of the pi bond and resonance stabilization.3 The resulting alcohol OH group can adopt either the R or S configuration. However, it is not possible to determine the relative amount of R and S product via 1H-NMR spectroscopy4. The R and S configurations are enantiomers, and enantiomers cannot be distinguished in an achiral environment, such as 1H-NMR. Consequently, the product must undergo further functionalization in order to be distinguishable via 1H-NMR spectroscopy.2 S-(+)-a-methoxyphenylacetic acid is added to the enzyme reduction product, forming a diastereomeric pair. The enzyme reduction product exists as both R and S, and the chiral acid is strictly S configuration. Diastereomers can be distinguished using 1H-NMR spectroscopy, and thus, the relative amounts of SR and SS isomer formed can be determined.4 The reaction scheme for the addition of S-(+)-a-methoxyphenylacetic acid to the enzyme reduction product is shown below.
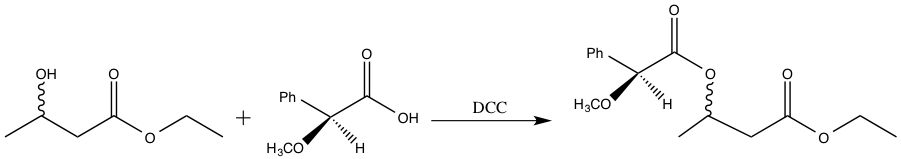
Figure 4: Addition of S-(+)-a-methoxyphenylacetic acid to the enzyme reduction product.
The reduction of b-ketoesters using baker’s yeast has been widely studied and is known to show enantioselectivity.5 Literature precedence supports the hypothesis that the S configuration will be favored over the R configuration.6
In terms of a “real world” application, reduction reactions involving baker’s yeast open a window of opportunity for “green chemistry.” Baker’s yeast is neither toxic nor pathogenic, and is a very economical catalyst.5 Additionally, by examining the varying strengths of the metal hydride reducing agents, selective reductions of carboxylic acid derivatives are possible.3
Experimental Procedure—Sodium Borohydride Reduction
The experimental procedure was very similar to the one outlined in the laboratory handout, with some minor modifications.1
4-tert-butylcyclohexanone (202 mg, 1.31 mmol, 1 equiv) was added to a 5 mL conical vial equipped with an air condenser, spinvane, and stir bar. Methanol (CH3OH) (200 mL, 4.94 mmol, 3.77 equiv) was added through the air condenser and the solution was stirred continuously. Sodium borohydride (NaBH4) (400 uL, 11.31 mmol, 8.56 equiv) was slowly added to the conical vial and the reaction was stirred for 20 minutes. Thin-layer chromatography was used to monitor the reaction. Thin-layer chromatography indicated the presence of starting material in the reaction mixture, and thus, the reaction was stirred for an additional 30 minutes. An infrared (IR) spectrum of the starting material was acquired while stirring.
Cold hydrochloric acid (HCl) (4 mL) was added drop wise to the reaction. The reaction was transferred to a 15 mL centrifuge tube. The aqueous layer was extracted via three 2 mL washes of methylene chloride (CH2Cl2). The centrifuge tube was lightly shaken to aid in the separation of the layers. The CH2Cl2 layer was extracted using a Pasteur pipette, and it was transferred to a drying column containing sodium sulfate anhydrous (Na2SO4). A tared 25 mL round-bottom flask was used to collect the dried eluates from the drying column. CH2Cl2 was used to rinse the remaining product from the drying column into the flask. A boiling stone was added to the flask. A sand bath was used to heat the flask and remove the CH2Cl2 from the reaction mixture. The reaction was concentrated to dryness, and the product was weighed. 1H-NMR and IR spectra as well as a melting point measurement of the product were acquired.
See attached page for calculations of mmol used, theoretical yield, and a flow chart for the “Work-up and Isolation of the Crude Product.”
Results/Data/Spectra
Table 1: 1H-NMR (CDCl3) Key Peak Table for 4-tert-butylcyclohexanone
|
Chemical Shift (ppm) |
Type of Signal |
J Values (Hz) |
Integration |
Type of Proton |
|
0.94 |
S |
—- |
9 |
CH3 |
|
1.76 |
M |
7.0, 7.0 |
4 |
CH2 |
|
1.77 |
M |
7.0, 7.1 |
1 |
CH |
|
2.22 |
T |
7.1 |
4 |
CH2 |
*Predicted using ChemBioDraw 13.0
Table 2: 1H-NMR (CDCl3) Peak Table for Hydride Reduction Product
|
Chemical Shift (ppm) |
Type of Signal |
J Values (Hz) |
Integration |
Type of Proton |
|
1.85 |
S |
—- |
9 |
CH3 |
|
3.45 |
T of T |
11.12, 5.16 |
2.39 |
CH2 |
|
4.15 |
Q |
5.82* |
0.99 |
CH2 |
*Average of all J-values, calculations shown on spectrum
Table 3: Infrared Spectroscopy Peak Table for 4-tert-butylcyclohexanone
|
Absorption (cm-1) |
Functional Group |
|
2946.85, 2867.14 |
Alkane C-H Stretch |
|
1720.59 |
Carbonyl C=O Stretch |
Table 4: Infrared Spectroscopy Peak Table for Hydride Reduction Product
|
Absorption (cm-1) |
Functional Group |
|
3292.91 |
Alcohol O-H Stretch |
|
2938.69, 2858.90 |
Alkane C-H Stretch |
|
1719.99 |
Carbonyl C=O Stretch* |
*Carbonyl peak transmittance: 81.36%
Melting Point Measurement: 45-530 C
Table 5: Class Data
|
Type of Hydride Reduction |
Product Ratio |
|
|
Sodium Borohydride |
2.4:1.0 (Trans : Cis) |
|
|
Lithium Aluminum Hydride |
9.5:1.0 (Trans : Cis) |
|
|
L-Selectride |
20:1 (Cis : Trans) |
|
|
Enzyme Reduction |
10% ee S; 44% ee S |
|
Discussion
The melting point measurement of the hydride reduction product was 45-530 C. The literature value for the melting point of the mixed isomers is 62-700 C.7 The melting point is a rather broad range, and occurs well below the literature value, indicating the product is not entirely pure. The product is a mixture of the cis and trans isomers as well as the starting material. Thin-layer chromatography and IR spectroscopy also indicate the presence of starting material and will be discussed further.
IR spectroscopy is particularly useful in determining the functional groups in a compound, and thus, is incredibly helpful in determining whether the reduction product formed. The IR spectrum of the starting material contains a characteristic peak at 1720.59 cm-1, representing a carbonyl C=O.3 In the IR spectrum of the hydride reduction product, this peak is still present, but not as intense (see “Table 4”). This indicates that there is starting material present in the product, and this is in agreement with the thin-layer chromatography analysis (see “Thin-Layer Chromatography”). The thin-layer chromatography plate indicated starting material was present in the reaction mixture, and thus, the reaction did not reach completion. The IR spectrum of the hydride reduction product exhibits a broad peak at 3292.91 cm-1, indicating the presence of an alcohol O-H.3 The appearance of this stretch supports the idea that the hydride reduction product was formed. However, the presence of the carbonyl C=O stretch in this spectrum shows that the product is not entirely pure, as it contains a significant amount of starting material.
The major product formed in this reaction was the trans-4-tert-butylcyclohexanol product. Upon analysis of the 1H-NMR spectrum, it is evident that the trans product formed over the cis product in a 2.4:1 ratio (see “Table 5”). The trans to cis ratio was determined by analyzing the 1H-NMR spectrum in the 3.0 ppm to 5.0 ppm region. In this region, there is a triplet of triplets at 3.45 ppm and a quintet at 4.15 ppm (see “Hydride Reduction Product 1H-NMR Spectrum). The integration values for each peak can be used to determine the relative ratio of trans to cis product. However, the two peaks must first be identified as either the trans peak or cis peak in order to determine the ratio.
In order to determine which peak corresponds to which isomer, the dihedral angles of the neighboring carbon-six and carbon-two protons in relation to the carbon-one proton must be investigated. Drawing a Newman projection of the trans product looking down carbon-two to carbon-one shows dihedral angles of 600 between H1 and H2 and 1800 between H1 and H2’. Therefore, the H2 and H2’ protons would both split the H1 proton, creating a doublet of doublets. Drawing a Newman projection of the trans product looking down carbon-one to carbon-six shows dihedral angles 600 between H1 and H6 and 1800 between H1 and H6’ Consequently, the H6 and H6’ protons would both split the H1 proton, creating a doublet of doublets. The H1 is split into a doublet of doublets of doublets of doublets, or more simply put, a triplet of triplets.8 The peak at 3.45 ppm is clearly identified as a triplet of triplets with a small amount of peak overlap. The Newman projections are shown in Figure 5 below.

Figure 5: Newman projections for the trans sodium borohydride reduction product. Looking down carbon two-carbon one (left) and carbon one-carbon six (right).
Drawing a Newman projection of the cis product looking down carbon-two to carbon-one shows dihedral angles of 600 between H1 and H2 and 600 between H1 and H2’. Drawing a Newman projection of the cis product looking down carbon-one to carbon-six shows dihedral angles 600 between H1 and H6 and 600 between H1 and H6’. Being that all four neighboring protons have the same dihedral angle to the H1 proton, they are equivalent. Consequently, these four protons create a quintet, and this is identified as the peak at 4.15 ppm.8 The Newman projections are shown in Figure 6 below.

Figure 6: Newman projections for the cis sodium borohydride reduction product. Looking down carbon two-carbon one (left) and carbon one-carbon six (right).
After identifying the peak at 3.45 ppm as the trans product and the peak at 4.15 ppm as the cis product, the relative ratio of trans to cis product can be calculated utilizing the integration values of the peaks. By dividing the integration for the trans peak by the integration for the cis peak, the relative ratio of trans to cis product was determined to be 2.4:1.0.
By examining the chair conformations of the cis and trans products, it is evident that the trans product is thermodynamically favored and the cis product is kinetically favored. It is clear that the trans product was formed in the sodium borohydride reduction of 4-tert-butylcyclohexanone. The thermodynamically favored product was formed because sodium borohydride is not a sterically hindered reducing agent, and is therefore able to attack the 4-tert-butylcyclohexanone from the top face of the molecule despite the presence of the large tert-butyl group. The addition of the hydride from the top face places the alcohol OH group in the equatorial position, forming the more energetically stable product.3 If the sodium borohydride attacked from the bottom face, the alcohol OH group would be forced into an energetically unfavorable axial position. In moving to this axial position, the oxygen must past through a highly unstable eclipsed conformation.4 Thus, the thermodynamically favored product is formed when using sodium borohydride in the reduction of 4-tert-butylcyclohexanone.
Lithium aluminum hydride selectively reduced the 4-tert-butylcyclohexanone to the trans product over the cis product in a 9.5:1.0 ratio. Lithium aluminum hydride is similar to sodium borohydride in that it is not sterically hindered and has the ability to the attack the carbonyl from the top face.4 Thus, when the hydride attacks from the top face, the alcohol OH group is pushed to the equatorial position. This forms the thermodynamically favored product. On the contrary, L-selectride selectively formed the cis product over the trans product in a 20:1.0 ratio. L-Selectride is a bulky, sterically hindered reducing agent, and thus, does not have the ability to attack from the top face of the starting material.4 It is forced to attack the 4-tert-butylcyclohexanone from the bottom face due to the presence of the bulky tert-butyl group. The bottom-ide attack pushes the alcohol OH group into an energetically unfavorable axial position. Although this product is not energetically favorable, it forms more quickly, and thus, is the kinetic product. Thus, the L-selectride reduction of 4-tert-butylcyclohexanone is under kinetic control.
The enzyme reduction reaction favored the S configuration product in 10% enantiomeric excess and 44% enantiomeric excess. The ethyl acetoacetate contains both a ketone and an ester functional group. The baker’s yeast selectively reduces the ketone to an alcohol instead of reducing the ester. The ester is more difficult to reduce due to the presence of the pi bond and resonance stabilization.3 Baker’s yeast is a naturally occurring chiral resolving agent and is stereoselective in the reduction of the ketone to an alcohol.2 It favors the S configuration, as demonstrated in the data. However, it is not possible to distinguish between enantiomers in an achiral environment, such as in 1H-NMR spectroscopy, and thus, the enzyme reduction product needed to be further functionalized to determine whether the R or S configuration was favored in the ketone reduction.4 A chiral acid, S-(+)-a-methoxyphenylacetic acid, was added to the enzyme reduction product. The enzyme reduction product exists as either R or S, and the chiral acid is strictly S. Thus, a diastereomeric pair was generated, which was easily distinguished via 1H-NMR spectroscopy. By analyzing the 1H-NMR spectrum, it was clear that the SS diastereomer formed over the SR diastereomer, and thus, the S configuration of the alcohol was favored in the enzyme reduction of ethyl acetoacetate.
Conclusion
The purpose of this experiment was to investigate the stereoselectivity of various hydride reducing agents.1,2 The reduction of 4-tert-butylcyclohexanone using sodium borohydride, lithium aluminum hydride, and L-selectride was studied in detail. After extensive analysis of 1H-NMR spectra, IR spectra, and melting point measurements, it is clear that sodium borohydride favored the trans product over cis product in a ratio 2.4:1.0, lithium aluminum hydride favored the trans product over cis product in a ratio of 9.5:1.0, and the L-selectride favored the cis product over trans product in a ratio of 20:1.0. The smaller, less bulky sodium borohydride and lithium aluminum hydride attacked the carbonyl carbon from the top face of the molecule, forming the trans and thermodynamically-controlled product.3,4 On the contrary, the large, bulky L-selectride attacked the bottom face of the molecule, forming the cis and kinetically-controlled product.3,4 The steric hindrance created by the bulky tert-butyl group did not affect the top-side attack of sodium borohydride and lithium aluminum hydride, but forced a bottom-side attack with the L-selectride. Thus, the hypothesis was correct.
The reduction of ethyl acetoacetate using baker’s yeast favored the S configuration in 10% enantiomeric excess and 44% enantiomeric excess. Therefore, it is evident that baker’s yeast favored the S configuration, and thus, the hypothesis was correct.
Future work could include 13C-NMR spectroscopy to further characterize the product, as well as proton-decoupling or COSY experiments to further analyze the 1H-NMR spectra.
References
- “Hydride Reductions of 4-tert-butylcyclohexanone,” Dr. Lynn Bradley, The College of New Jersey.
- Patterson, J.; Sigurdsson, S. “Use of Enzymes in Organic Synthesis: Reduction of Ketones by Baker’s Yeast Revisited,” J. Chem. Educ. 2005, 1049-1050.
- Brown, William, Christopher Foote, Brent Iverson, and Eric Anslyn. Organic Chemistry. 5th ed. Brooks/Cole, 2008. Print.
- “Lecture Notes on Reduction Reactions,” Dr. Lynn Bradley, The College of New Jersey.
- Mahmoodi, N. O.; Navrood, M. N. “Enantio-, regio-, chemoselective of aromatic a-diketones by baker’s yeast in diverse organic-water solvent systems,” Arkivoc. 2007, 37-45.
- “Experiment 8, Synthesis and Purification of Diastereomers: Sodium Borohydride Reduction of a Chiral Ketone,” Dr. Alan Shusterman, Reed College.
- Mayo, Pike, and Trumper. Microscale Organic Laboratory. 3rd ed. New York: John Wiley and Sons, 1994. 165. Print.
- “Lecture Notes on 1H-NMR Splitting,” Dr. Lynn Bradley, The College of New Jersey.
