Multi-step Synthesis of Ketal Ester
Written by Jimmy
Table of 1H NMR Spectral Data of Ketal ester
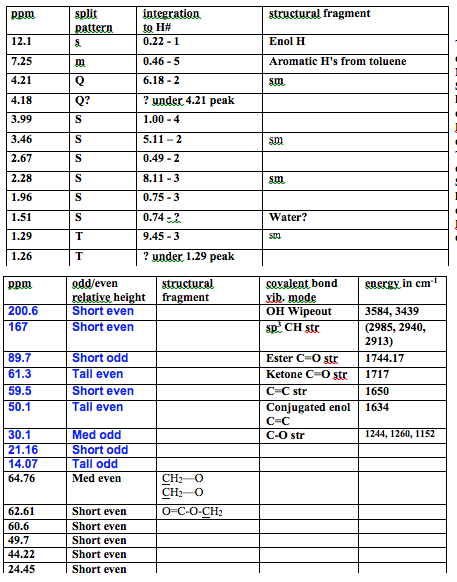 Table of 13C NMR Spectral Data of Ketal ester Table of IR Spectral Data of Ketal ester
Table of 13C NMR Spectral Data of Ketal ester Table of IR Spectral Data of Ketal ester
Provide your experimental index of refraction and correction factor: 1.4229 @ 22ºC
1.4229+0.00045*2=1.4238 Literature=1.4377
Reproduce your Theoretical and Percent Yield Calculations.
Ethyl Acetoacetate: 10.0 mL (1.029g/mL) (1/130.14g/mol) = 0.0791 mol
Glycol: 4.5 mL (1.113g/mL) (1/62.07g/mol) = 0.0807 mol
1:1 ratio of prods to reacts and reacts to reacts
Ethyl Acetoacetate is limiting
.
Theoretical yield Percent Yield
Product: 174 g/mol (0.0791 mol)= 13.76g 2.42g/13.76g *100 = 17.6% (if it were pure)
Water: (0.0791 mol)(18.0 g/mol)(1/1.00mL/g)=1.425
GC/MS results
| Retention Time (min) | % | Structure |
| 3.652 | 19.47 | |
| 6.498 | 68.17 | |
| 9.992 | 12.36 |
Table of 1H NMR Spectral Data of Alcohol
Ketal Ester
Table of 13C NMR Spectral Data of Alcohol Table of IR Spectral Data of Alcohol
Provide your melting point and voltage: 165.4-167.1 °C @ 47 volts.
Reproduce your Theoretical and Percent Yield Calculations.
PhBr: 9.0 mL (1.491g/mL) (1/157.01g/mol) = 0.08547 mol (2.33 eq)
Mg: 2.00 g (1/24.31 g/mol) = 0.08224 mol (2.24 eq) LR of gringard formation
Methyl Benzoate: 5.00 g (1/136.15 g/mol) = 0.03672 mol (1 eq) overall LR (despite 2:1 ratio)
2:1 ratio of gringard to ester. 1:1 ratio of ester to product.
.
Theoretical yield Percent Yield
0.03672 mol (1 mol ester/1 mol alcohol)(260.3 g/mol) = 9.56 g 3.405 g/9.56 g *100 = 35.6%
Results/Discussion:
Ethyl acetoacetate was reacted with ethylene glycol by an acid catalyzed nucleophilic addition across the ketone’s carbonyl using p-toluene sulfonic acid (pTsOH) in toluene to form a diether ketal ester will be reacted further with two equivalents of phenylmagnesiumbromide (which was formed by reacting phenyl bromide with magneisum) by nucleophilic substitution/addition, which was worked up in acidified water to yield ethyl acetoacetate ethylene acetal. For the acid catalyzed addition, ethyl acetoacetate, toluene and a speck of pTsOH were added to a Round bottom flask (RBF) to yield a clear solution. Ethylene glycol was added and clear, beady liquid formed at the bottom of the solution showing that ethylene glycol is not soluble in toluene. The solution was set to stir and reflux, and after the reflux the solution remained clear and colorless, but the bottom beady layer seemed to disappear indicating that the ethylene glycol should have created with the ethyl acetoacetate.
The solution was then distilled at a temperature of 104 ºC. Initially all the solution coming over was one phase and cloud this is not good because it should be forming a water co-product and coming over as a bi-phasic solution. Eventually the bi-phasic solution of water and toluene began to come over and be collected, indicating the reaction was proceeding and forming the water co-product. The distillate was gray and cloudy indicating possible starting material/product coming over. The distillation was stopped when the solution being distilled seemed clear and free of water, despite not getting the anticipated amount of water co-product collected. The solution was then washed using NaOH to neutralize the acid, and water to remove any left over salts from the acid/base. This was capped and dried over salts overnight.
The 1H NMR is very messy and indicates a majority of starting material. The starting material peaks are as follows. There is a quartet at 4.21 ppm with integration of 2 corresponding to the hydrogens attached to the carbon attached to the oxygen. There is a singlet at 3.46 ppm with integration of 2 corresponding to the hydrogens attached to the carbon in between the carbonyls. There is a singlet at 2.28 ppm with integration of 3 corresponding to the hydrogens of the methyl off of the ketone carbonyl. There is a triplet at 1.29 ppm with integration of 3 corresponding to the hydrogens on the methyl of the ethyl chain. These all match literature values which are in order of the peaks listed above: Q 4.202, S 3.451, S 2.273, T 1.28.1 There also seems to be a slight toluene contamination with peaks that fall at 7.31-7.18 ppm which matches the literature value of 7.38-7.001. There is also a small peak at 12.11 corresponding to the enol H, which shows there is a lot of starting material. There is also an unknown impurity at 1.51 ppm which could possibly be water. As for the product, there is supposed to be a quartet at 4.18 ppm, but it is hidden under the starting material peak which makes the integration impossible to find, it corresponds to the hydrogens attached to the carbon attached to the oxygen. There is a singlet at 3.99 ppm with integration of 4 corresponding to the hydrogens attached to the carbons in the ether ring. There is a singlet at 2.67 ppm with integration of 2 corresponding to the hydrogens attached to the carbon in between the carbonyl and the carbon attached to two oxygens. There is a singlet at 1.96 ppm with integration of 3 corresponding to the hydrogens of the methyl off of the carbon attached to two oxygens. There is supposed to be a triplet at 1.26 ppm, but it is under starting material peaks, which makes the integration impossible to find, corresponds to the hydrogens on the methyl of the ethyl chain. The integration is 5:1 starting material to product. This shows the reaction was not successful and most of the substance is starting material, with some toluene impurity.
The 13C was no better than the 1H NMR. There is mostly starting material, so much, that the enol form is visible as well. The short even ketone peak is at 200.64 ppm. There is a short even ester peak at 167 ppm from the keto form. The CH carbon of the C=C in the enol has a short odd peak at 89.74 ppm. The carbon single bonded to one oxygen in the keto form has a tall even peak at 61.34 ppm. The same carbon, but in enol form has a short even peak at 59.88 ppm. The carbon in between the carbonyls has a tall even peak at 50.11 ppm. The methyl carbon attached to a carbonyl has a medium odd peak at 30.11 ppm. That same carbon in enol form has a short odd peak at 21.16 ppm. The methyl carbon of the ethyl chain has a tall odd peak at 14.07 ppm. All of the peaks match literature value within 0.1 ppm1. Some of the enol peaks cannot be seen because it was too dilute; however, being able to see any enol shows there was mostly starting material. All of the product peaks are not visible because there was not enough formed to be concentrated enough. There is a medium even peak at 64.76 ppm that corresponds to the symmetric carbons in the CH2‘s of the ether ring. The carbon with two oxygens is not visible, but it should be around 100 ppm because there are two oxygens deshielding it. Also the ester peak is likely under the peak at 167 ppm. There is a short even peak at 62.61 ppm corresponding to the carbon single bonded to the oxygen of the ester and the methyl. There are 2 extra unknown short even peaks at 60.5 ppm and 49.7 ppm. There is a short even peak at 44.22 ppm corresponding to the carbon of the CH2 between the carbonyl and the ether ring. There is a short odd peak at 24.45 ppm corresponding to the carbon of the methyl attached to the ether ring. The other methyl in the product does not have a visible peak. This agrees with the proton NMR in that almost all of the material is starting material.
The IR spectrum matches almost perfectly with the literature IR data for ethyl acetoacetate.1 There are absolutely no peaks from the product aside from a possible sharp peak at 3583.95 cm-1, which could just be an enol H. I have OH stretching at 3439 cm-1 matching literature of 3438 cm-1 . I have sp3 CH stretching at 2985 cm-1 , 2940 cm-1 , and 2913 cm-1 , and literature data is 2985 cm-1 , 2941 cm-1 , and 2910 cm-1 . The C=O ester peak is at 1744 cm-1, with lit at 1731 cm-1. The C=O ketone peak is at 1717 cm-1, with lit at 1719 cm-1. The C=C strs for the enol form come at 1650 cm-1 and 1634 cm-1 and with lit at 1651 cm-1 and 1634 cm-1. The fingerprint regions match exactly in looks and all of the peaks are within +/- 3 cm-1 of observed data. No literature value peaks for toluene are present in the IR due to it’s low concentration, which is also why there are no product peaks. This adds to the all of the data suggesting there is almost all starting material present.
The GC/MS data agrees with the carbon and proton NMR. There is a peak with retention time of 3.652 and 19.47 % area that corresponds to toluene. There is a peak with retention time of 6.498 and 68.17 % area that corresponds to unreacted ethyl acetoacetate. There is a peak with retention time of 9.992 and 12.36 % area that corresponds to unreacted the product ethyl acetoacetate ethylene acetal. This confirms that the material is majority starting material.
The index of refraction was1.4229 @ 22ºC, this was corrected to 1.4238. The literature value for the product is 1.4377. The literature value for ethyl acetoacetate is 1.41922. The observed refractive index was low, which is consistent with having starting material in it. This also agrees that the product is impure.
See conclusion and mass from the first paper because my file got deleted
For the methyl benzoate and gringard, silver Mg was ground in mortal in pestal and put in a RBF with clear diethyl ether. This was attached to a dry reflux set up and addition funnel. Clear bromobenzene in diethyl ether was put in the addition funnel. I2 was added to the RBF, which turned it yellow, this was to promote gringard formation. A splash of the PhBr was added to start the gringard formation reaction. The mixture turned grey and more PhBr was added dropwise. The mixture began to fizz and turned light brown, which indicates formation of the gringard. The PhBr was continually added dropwise while the solution refluxed. The mixture turned dark brown, and there was noticeably less Mg, both of which indicate formation of the gringard.
Methyl Benzoate in ether was added to the addition funnel and added dropwise to the mixture. The solution turned wine/red and cleared up, there was still Mg though. This indicates the reaction of the gringard and the methyl benzoate is taking place. This was refluxed which turned it opaque red/orange and precipitate formed showing that the gringard reaction has successfully taken place. This was cooled and quenched with H2SO4 and NaHSO3. This created a lot of bubbling showing the Mg reacting with the acid to form the Mg salts. This yielded a solution with a clear yellow layer on top and a cloudy white/yellow translucent layer on bottom.
An extraction series of H2SO4 (to remove Mg salts), sat’d NaCl (to remove acid), NaHCO3 (to neutralize trace acid), and sat’d NaCl (to remove water) to yield a slightly cloudy light yellow solution. This was dried over salts which cleared the solution up. After drying the solution was rotovapped half way down until white crystals formed in solution. This was chilled to drop more crystals, and then filtered with a hirsch funnel and a ligroin rinse. The filtration yielded white shiny crystals which were dried between filter paper. A lot more crystals formed in the filtrate, but there were plenty for analyzation, so they were left. The crystals collected were analyzed using melting point, IR spec, and 13C and 1H NMR.
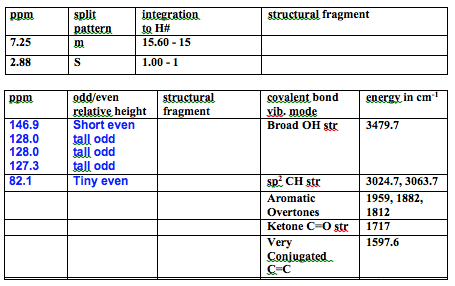
The 1H NMR had 2 peaks, one multiplet at 7.25 ppm with integration of 15 corresponding to all of the aromatic H’s in the benzene which deshielded to the same position. There is also a singlet at 2.88 ppm with integration of 1 corresponding to the alcohol hydrogen. There were no more peaks which means the product is pure.
The 13C NMR had a few more peaks. There were aromatic peaks consistent with mono-substituted benzene rings (146.98 ppm – short even, 128.07 ppm – tall odd, 128.04 ppm – tall odd, 127.36 ppm – medium odd). There is also a tiny even peak at 82.16 ppm which correlates to the carbon attached to the hydroxy and benzene rings. All this data matches the literature within 0.3 ppm.1 There were no other peaks which agrees with the proton NMR in that the product is clean.
The IR data is consistent with the product. There is a broad OH str at 3479.7 cm-1. There are sp2 C-H strs at 3053.7 cm-1 and 3024.7 is where chloroform comes in, but there are probably some under it because literature source has a peak at 3034cm-1.1 There are aromatic overtones at 1959, 1882, and 1812 cm-1. There is a C=C str at 1597.6 cm-1 corresponding to the C=C in the benzene rings. Finally there is a C-O str at 1180 cm-1 because the peak at 1216 cm-1 is from chloroform. My IR is very concentrated, but all of the peaks match the lit within 5 cm-1 except the C=C which is at 1583, but it is close enough.1 All this data is consistent with the intended product.
A melting point was run at 47 voltage and found to be 165.4-167.1 °C, this is not broadened out 2 degree window, but it is out of the error of +/- 3 degrees from literature (160-163 °C). The melt-temp has seemed to run high lately, so that is likely the reason for the elevated melting point. Therefore, this data agrees with all the other data that the product is pure.
The mass recovery was 3.405 g which is 35.6 % yield. Yield was so low because so many crystals formed and were left in the filtrate. At the beginning not all of the Mg did not react, so the reaction did not go to completion for the gringard formation. Also when I came back to do the work up the next week, my RBF was on its side and some may have came out. Additionally, the mixture could have been rotovapped a bit more to drop more crystals.
For the triphenylmethanol all of the data agrees that the product was pure. The yield was low, but mostly because the crystals in the filtrate were not collected.
Overall, the first reaction to make the ketal ester yielded almost all unreacted starting material, and in low yield, so that product and reaction scheme was given up on. Instead methyl benzoate was reacted phenyl magnesium bromide to yield 3.405 g of white shiny triphenylmethanol crystals. The product was pure, but in low yield due to the lack of collection of crystals in the filtrate.
references:
1) http://sdbs.riodb.aist.go.jp/sdbs/
2) http://www.sigmaaldrich.com/
