pGLO Lab Analysis
pGLO Lab
By: Yi Yu
Abstract
Plasmids are pieces of circular DNA that are in bacteria which can code for genes that can cause the bacteria to have extra “features”. With the pGLO plasmid this extra “feature” causes bacteria to glow (from the Green Fluorescent Protein that was inserted) and also has resistance to ampicillin (from the beta-lactamase protein). By adding the pGLO plasmid the E.coli bacteria will be able to grow on agar with arabinose sugar and ampicillin. The main reason for doing this lab is to better understand how genes control traits, they best way to do this is by doing mutagenesis. There are a few different ways to preform mutagenesis and in this lab it is done by adding a transposon to the pGLO plasmid and then looking at how the transposon effects how the bacterium looks and grows. By looking at how the bacteria grows and looks we can determine where the transposon inserted; this helps us better understand how genes control traits. In the sample we worked with the mutagenized bacteria did not glow green under UV light which tells us that the transposon inserted into the araC gene, and is confirmed in the electrophoresis gel.
Introduction
Plasmids have been studied in genetic research since they were discovered in 1952 by Joshua Lederberg. They have been used to figure out how certain bacteria reacts to different types of antibiotics and to change how bacteria functions by adding things into the plasmids. In this lab the plasmid that we focused on was the pGLO plasmid. This plasmid has three main coding regions that we will be looking at. The first gene region is the region that encodes for the beta-lactamase (bla) enzyme. “The beta-lactamase protein is produced and secreted by bacteria that contain the plasmid. Beta-lactamase inactivates the ampicillin present in the LB nutrient agar, allowing bacterial growth. Only transformed bacteria that contain the plasmid and express beta-lactamase can survive on plates that contain ampicillin.” (Leatherman 2011). The Green Fluoresce Protein (GFP) is a gene code that is inserted into the pGLO plasmid to make it glow green under UV light with the help of the araC gene. The araC gene encodes a protein that binds to arabinose and the promoter region as well in the presence of arabinose, when bound to the promoter region of the GFP gene it translates the protein that will make the bacteria glow green.
Mutagenesis does not happen very often in nature but can be used regularly in genetics labs to help study different functions of bacteria and plasmids. By doing mutagenesis on bacteria we can better understand how bacteria translate genes and how they react to their environment when something is changed. There are a couple ways to preform mutagenesis like using chemicals or X-rays. In this experiment mutagenesis is performed by inserting a transposon into the pGLO plasmid. The Tn5 transposon will be inserted into the pGLO gene; the Tn5 transposon encodes for a protein that leads to resistance of kanamycin, which will help us see if the transposon inserted correctly.
After the transposon is inserted into the pGLO plasmid, it will be taken up by E.coli bacteria through transformation. Transformation is when bacteria take in free DNA from their surroundings. Once the plasmids were taken in the bacteria was grown on nutrient agars with different combinations of ampicillin, kanamycin, and arabinose along with control groups to see how the mutant bacteria was affected by the transposon.
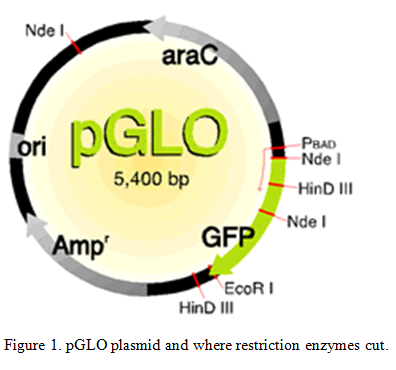
Bacteria use restriction enzymes on their own to get rid of bacteriophages by either cutting the proteins that they make or by cutting the phage directly (Aude). We are able to use these restriction enzymes to do restriction enzyme digest to cut the plasmids that were inserted into the bacteria. “Restriction enzymes are commonly used to cut DNA at particular nucleotide sequences. Often this is to make a new “recombinant piece of DNA, and is followed by ligating two pieces together.” (Leatherman 2011). The plasmids were cut at known locations of the plasmid with Nde 1 and EcoR1 enzymes. Knowing the amount of base pairs that are in the plasmid (5,400 bp) and where the restriction enzymes cut we can examine the different pieces of the cut plasmid to find out where the transposon inserted.
Methods
Use the Tn5 insertion kit to insert the Tn5 transposon into the pGLO plasmid. The first step was to mix 6.4 microliters of water, 1 microliter of EZ Tn5 10X reaction buffer, 1 microliter of .2 micrograms/microliter of pGLO plasmid, 0.6 microliters of Tn5 transposon, and 1 microliter of EZ-Tn5 transposase (helps transposon insert) in this order and mix by vortexing, then centrifuge. Incubate tube at 37 degrees Celsius for 50 minutes, then take tube out and spin again and return tube to incubator for another 50 minutes. Spin once more and add 1 microliter of stop solution to tube and mix well and spin liquid to bottom of the tube again. Heat tube to 70 degrees for 10 minutes; store in freezer for next time.
Transfer 1.5 mL of E.coli to three microcentrifudge tubes and put on ice for 10 minutes. Centrifuge tubes for 1 minute and get rid of all supernatant. Add 300 microliters of ice-cold CaCl2 to all three tubes; dissolve the pellet completely by pipetting and place tubes on ice for 2 minutes. Centrifuge tubes for 1 minute and discard the supernatant. Add 60 microliters of CaCL2 to each tube again and gently re-suspend the pellet and put on ice for 10 minutes. Label the three tubes, No DNA, pGLO wt, and pGLO mutant. Add nothing, 1 microliter of pGLO DNA, and 1 microliter of Tn5 mutagenesis pGLO reaction to each tube respectively. Gently mix tubes by tapping them and place on ice for 15 minutes. Heat shock the tubes at 42 degrees for exactly 90 seconds and return to ice immediately for 2 minutes. Add 1 mL of LB broth to tubes then transfer contents of each tube to a test tube and incubate at 37 degrees for 30 minutes with shaking. Obtain and label 5 different agar plates like shown in table 1 and label them. Transfer bacteria into new microcentrifudge tubes and spin them for 1 minute and remove 500 microliters of supernatant, re-suspend bacteria in broth. Add 200 microliters of each reaction to their respective agar plates and spread bacteria over the agar plates. Incubate plates at 37 degrees in prep room for a day.
|
DNA |
Type of plate |
Prediction |
| No DNA | Nothing added | Lawn of white |
| No DNA | Ampicillin | No growth |
| pGLO wild-type | Ampicillin | White colonies |
| pGLO wild-type | Ampicillin + Arabinose | Green colonies |
| pGLO mutagenized | Kanamycin + Arabinose | Green + White colonies |
Table 1. DNA added to the different plates and predictions.
Prepare two sterile culture tubes with 5 mL of LB broth, one with ampicillin and one with kanamycin, label which is which. Examine plates and choose on wild-type colony and put colony into the LB broth with ampicillin. Look at plate with the pGLO mutant and look to see if any of the colonies glow green under UV light. Colony that was picked for this experiment was white. Take colony and put in LB broth with kanamycin. Put tubes into incubator at 37 degrees and incubate overnight. Look at all plates and compare the results with your predictions. Get cultures from the incubator and transfer them into 15 mL tube to centrifuge. Spin in refrigerated centrifuge at 3000 rpm for 5 minutes and discard supernatant. Add 250 microliters of P1 buffer (removes RNA) to each bacteria pellet and re-suspend the pellet, and immediately transfer to microcentrifudge tube. Add 250 microliters of P2 buffer (lysis of cells) and mix by inverting for no more than 5 minutes. Add 350 microliters of N3 buffer (pH ideal for column binding) and mix immediately by inverting the tube. Spin for 10 minutes until there is a white pellet that forms. Pipet supernatant into separate spin columns and spin for 30 seconds then discard flow-through. Add 750 microliters of PE buffer (washes DNA) to column and spin for 30 seconds and discard flow-through. Place column in new 1.5mL microcentrifudge tube; add 50 microliters of water (elution of DNA) to spin column and let it stand for 1 minute. Spin for 1 minute then discard spin column and keep the flow-through. Measure the concentrations of the two samples of DNA, then put in freezer for next week. Concentration of wild-type DNA was 68.2 nano grams/microliter with a purity of 1.84 and the mutant DNA had a concentration of 81.9 nano grams/microliter with a purity of 1.64 which was not as pure
Use the restriction map to determine the number of bands in the wild-type plasmids cut at the different places.
| Uncut | Nde1 | EcoR1 | Nde1 + EcoR1 | |
| Number of bands and their sizes | 5,400 bp | 3,300 bp1,800 bp
300 bp |
5,400 bpNot circular | 2,800 bp1,800 bp
500 bp 300 bp |
Table 2. Band sizes of wild-type plasmids with different cuts.
Mix each solution based on what each tube needs. Use the concentration of DNA to determine how much DNA needs to be added to each tube (500/concentration=how much DNA is need).
| Uncut wt | Uncut mutant | Nde1 wt | Nde1 mutant | EcoR1 wt | EcoR1 mutant | Nde1 + EcoR1 wt | Nde1 + EcoR1 mutant | |
| Water | 12.67 | 13.9 | 10.17 | 11.4 | 10.17 | 11.4 | 10.12 | 11.4 |
| 10X buffer | None | None | 2Buffer 4 | 2Buffer 4 | 2 EcoR1 buffer | 2 EcoR1 buffer | 2 EcoR1 buffer | 2 EcoR1 buffer |
| DNA: 5 milligrams | 7.33 | 6.1 | 7.33 | 6.1 | 7.33 | 6.1 | 7.33 | 6.1 |
| Nde1 enzyme (20units/microliter) | None | None | .5 | .5 | None | None | .5 | .5 |
| EcoR1 enzyme (20units/microliter) | None | None | None | None | .5 | .5 | .5 | .5 |
Table 3. Mixtures of each tube. (all in microliters)
Place the eight tubes in incubator at 37 degrees for 1.5 hours for reaction to proceed, once 1.5 hours is up put tubes in freezer for next time.
Dilute 50X buffer to 1X buffer; add 20 mili liters of 1X TAE buffer in a graduated cylinder and then add 980 mili liters of water to the cylinder. Make a 1.2% agarose gel; weigh out .6 grams of powder agarose and empty into a flask, then take 50 mL of 50X TAE buffer and add it to the flask. Mix the agarose and the buffer then put it into a microwave for about a minute or until the solution becomes clear. Let the flask cool until it you can touch the flask without burning yourself. Set the tray in the running box with both side up and the comb in place and pour the agarose solution into the tray. Let the agarose sit until it becomes a milky white color and hardens. While waiting load 2 microliters of 10X loading dye to each tube and mix by pipetting. Lower the sides of the tray and then fill the running box with the TAE buffer until it covers the top of the gel. Load the 1kb ladder on the far left end with 10 micro liters of 1kb ladder. Take the PCR reactions from the previous week and fill in the next 8 spaces in the gel with 20 micro liters of the restriction digest reactions in order. After the gel is loaded run the gel for about 45 minutes at 100 volts, check periodically to see if the dyes are moving down the gel. Once the dye has gone about half way down the gel take it out of the running box and place the gel in a staining box. Pour SYBR green into the staining box until the SYBR green covers the top of the gel, swirl the box every 5 minutes. The SYBR green binds to the DNA and allows us to look at the DNA under a UV light. After 15 minutes take the gel out of the box and pour the SYBR green back into its bottle. Take your gel and place it in a UV box and take a picture of your gel to see the results of your PCR.
Results
When the bacteria was first examined the mutagenized bacteria did not glow green when put under UV light which means that the transposon could have inserted itself into either the araC gene or into the GFP gene.
|
DNA |
Type of plate |
Results |
| No DNA | Nothing added | Lawn of white |
| No DNA | Ampicillin | 13 colonies |
| pGLO wild-type | Ampicillin | White colonies over 100 |
| pGLO wild-type | Ampicillin + Arabinose | Green colonies over 100 |
| pGLO mutagenized | Kanamycin + Arabinose | 8 all white colonies |
Table 4. Results of the agar plates.
When mixing the restriction enzymes, the restriction enzyme was mistaken for buffer when being added to the mutant tubes which lead to the mutant tubes to be unreadable on the gel.
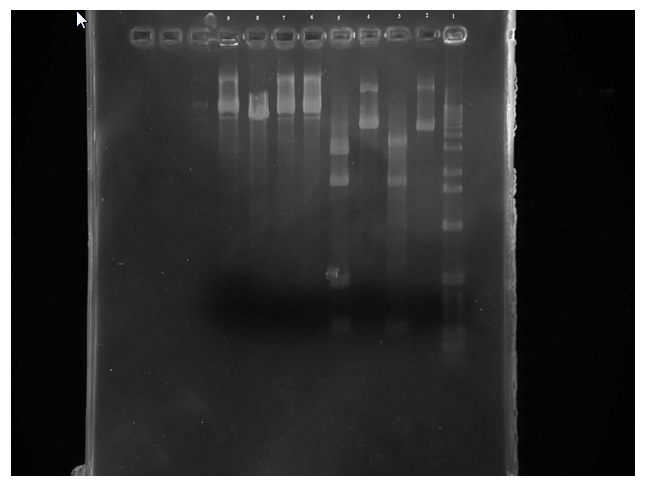
Figure 2. Row 1 is the 1kb ladder. 2 wild-type uncut plasmid. 3 wild-type nde cut. 4 wild-type eco cut. 5 wild-type eco/nde cut. 6 mutant uncut. 7 mutant nde cut. 8 mutant eco cut. 9 mutant eco/nde cut
From this gel we can see that the wild-type uncut and EcoR1 cut are the same which is what was expected. There are four bands on the wild-type EcoR1/Nde1 cut in the 300 and 500 regions and in the 1000 and 2000 areas. And three bands on the Nde1 cut in the 300, 1000, and 2000 areas. So all of the wild-type samples were where they were predicted to be. Unfortunately the mutant samples are not readable.
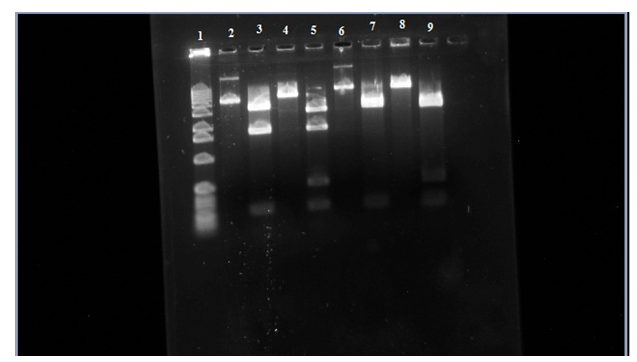
Figure 3. Row 1 is the 1kb ladder. 2 wild-type uncut plasmid. 3 wild-type nde cut. 4 wild-type eco cut. 5 wild-type eco/nde cut. 6 mutant uncut. 7 mutant nde cut. 8 mutant eco cut. 9 mutant eco/nde cut.
In this figure you can see that in row 6 it didn’t travel as far as the wild-type uncut in row 2 telling us that the transposon is in the sample. Looking at row 7 and 9 there is a missing bar at around 1,800 base-pairs like in rows 3 and 5. This is where the transposon is inserted itself into to the araC gene which is in one of the Nde1 cuts that it about 1,800 base-pairs. In the gel the addition of the transposon to this region causes the 1,800 bar to stay back to where the 2,800 base-pair bar is and makes it look like just one bar on the gel.
Discussion
The transposon was found in the araC gene of the pGLO plasmid from the gel results. With the transposon being in the araC gene it doesn’t make the protein that binds to the promoter region of the GFP gene which resulted in the mutagenized bacteria to not glow green under the UV light. The insertion of the transposon in the araC gene might also explain why there was so little mutant growth on the plate the mutant bacteria was placed on. The problem with the original gel was that more enzyme was added to the tubes which made the mutant samples unreadable on the gel.
References
Aude A Bourniquel, Thomas A Bickle, Complex restriction enzymes: NTP-driven molecular motors, Biochimie, Volume 84, Issue 11, 1 November 2002, Pages 1047-1059, ISSN 0300-9084, 10.1016/S0300-9084(02)00020-2.
Leatherman J. 2011 Genetics Laboratory Manual. University of Northern Colorado
