Mutation of the Eyeless Gene in Drosophila melanogaster
Written by Danielle
A mutation of the eyeless gene in Drosophila melanogaster changes the phenotype and is demonstrated to be located within intron 2 of the eyeless gene.
Abstract:
Drosopholia melanogaster flies are ideal for genetic study. This is due to the ability to observe mutation of the DNA through phenotypic manifestations. A mutation is an alteration in the DNA that may or may not alter gene expression within the organism containing the modified sequence. With use of D. melanogaster flies in genetic study, it was found that frequent progeny demonstrate phenotypic change that can be attributed to mutation. One such mutation was identified to modify the eyeless gene of D. melanogaster. The purpose of this study is to utilize D. melanogaster flies, selected for a mutation in their eyeless gene, to observe the phenotypic effect of such a mutation while employing techniques to further define the mechanism of phenotypic alteration. It was hypothesized that a mutation within the eyeless gene of D. melanogaster produces a phenotypic change in the organism, and through in-depth analysis of the eyeless gene sequence, dissimilarities in the mutant’s DNA can be utilized to propose mutational mechanism. Through DNA sequencing of the mutant eyeless sequence, the gene was localized to occupy a portion of exon 2, extending through intron 2, and a portion of exon 3 of the D. melanogaster wild-type eyeless gene. This confinement allowed for proposed placement of the mutation, promoting hypothetical mutational mechanism.
Introduction:
A mutation is an alteration in the DNA that may or may not alter gene expression within the organism containing the modified sequence. Gene expression effect is dependent on location and extensiveness of the change. For example, a mutation located within a noncoding portion of the DNA may not alter gene expression in a way that is visible through phenotype. On the other hand, a mutation within an enhancer, repressor, or gene sequence that codes for protein may have a significant effect on an organism’s phenotype. It is feasible that this change of the DNA be beneficial, harmful, or negligible in the organism as a whole. Because there are numerous ways in which a DNA sequence can be altered, several different classifications of mutation exist based on the change of sequence detected (1). This includes a substitution mutation in which one base is exchanged for another, an insertion or deletion mutation resulting in gain or loss of nucleotides, respectively, and the resulting frame shift effect of mutation, where the reading frame for codons encoding amino acid polymerization to form polypeptides is altered to disrupt translation (1). This frame shift effect is capable of influencing specific protein production, which in turn, often disrupts cellular signaling pathways, the most often observed mechanism for phenotypic change when DNA is altered.
When looking to investigate mutational mechanism from observed phenotype, it is vital to employ a model organism in which variation can be minimal and manipulation of genomic information is relatively simple. One such organism is Drosophila melanogaster. First illuminated in the work of F.W. Carpenter and William Castle, D. melanogaster is now one of the most widely used model organisms for studying principles of genetic inheritance (2). Following Carpenter and Castle’s effort, Thomas H. Morgan commenced experimentation that fostered the widespread popularity of D. melanogaster use (2). A short life cycle, small size, ease of use, and having only 4 pairs of chromosomes collectively make D. melanogaster advantageous in genetic lab work (3). Additionally, male and female D. melanogaster flies can be distinguished by size and color, further reducing unwanted variability. Males are smaller than females and contain darker banding on their ventral abdomen (3).
One factor that makes D. melanogaster so ideal for genetic study is the ability to observe mutation of the DNA through phenotypic manifestations. When D. melanogaster began to be used extensively in genetic studies, it was found that multiple phenotypes were observed in progeny. It was then seen that these changing phenotypes could be attributed to spontaneous mutation (4). One such spontaneous mutation was identified to modify the eyeless gene of D. melanogaster (4). Eyeless is located on the fourth chromosome and when mutated, appears to produce variability in the size of the eye (4). It was observed in previous experimentation that the eye of a mutant eyeless fly is not completely absent, but rather exhibits variable expressivity. The eye appears smaller than that of the wild-type fly. It has been shown that this variable expressivity can be influenced by environment and genetic factors (5). The eyeless gene is so influential in eye development that studies have demonstrated the ability for researchers to induce eye growth in abnormal locations simply based on the activation of the eyeless gene in those areas (6). It is the quality of being so effortlessly manipulated which makes eyeless superlative for mutational study.
The purpose of this experiment is to utilize D. melanogaster flies, selected for a mutation in their eyeless gene, to observe the phenotypic effect of such a mutation while employing techniques to further define the mechanism of phenotypic alteration. These techniques include observation of the polytene chromosomes encompassing the genetic material, followed by isolation of the genomic DNA, amplification of the sequence containing the mutated eyeless gene, cloning of that sequence into a self-replicating expression plasmid, sequencing of those plasmids containing the mutated eyeless gene, and analysis of the specific gene to distinguish the altered DNA from that of a wild-type fly. It is anticipated that differentiating the mutated DNA will allow for more explicit analysis of the mechanism inducing the mutation. It is hypothesized that a mutation within the eyeless gene of D. melanogaster produces a phenotypic change in the organism, and through in-depth analysis of the eyeless gene sequence, dissimilarities in the mutant’s DNA can be utilized to propose mutational mechanism.
Materials and Methods:
Cultures of wild type and eyeless flies were treated in the manner discussed in the Cold Spring Harbor Protocol for “Culture of Drosophila” (7) (Ward’s Natural Science, San Luis Obispo, CA).
Polytene Chromosomal Squash Prep
To determine if a mutation in the eyeless gene could be observed at a chromosomal level, the salivary glands from third instar larva were dissected out and the polytene chromosomes within them were isolated. The larva were allowed to incubate in a 0.7% NaCl solution prior to dissection. Post-dissection the salivary glands were transferred to 45% acetic acid and allowed to incubate for 10 minutes. The chromosomes were then stained with 2% Aceto-Orcein stain (Carolina Bio Supply, Gladstone, OR) After a 10 minute incubation, the stain was removed with a 45% acetic acid wash done 3 times. The chromosomes were then transferred to a microscope slide, immersed in water, and covered with a coverslip. Excess stain was removed with Whatmann paper and the chromosomes were viewed under a compound microscope. Both wild type and eyeless mutant third instar larva were used for comparison.
Genomic DNA Isolation
Thirty wild-type and thirty mutant eyeless flies were anesthetized using FlyNap Anesthetizer (Carolina Bio Supply, Gladstone, OR). These flies were transferred to a microcentrifuge tube and spun at 13 xg for 1 minute. Genomic DNA was extracted from both wild-type and eyeless mutant adult flies using the protocol described in the Wizard® SV Genomic DNA Purification Kit (Promega, USA). The protocol was modified so that RNAase was not included in the lysis mix. A 1% agarose gel was run for 30 minutes at 85 mA to verify the extraction. Twenty-five micro-liters of wild-type and eyeless sample were loaded into the gel, respectively. Twenty microliters of 1 kb DNA ladder was loaded into the first lane of the gel, before the gel apparatus was covered and allowed to run.
PCR of the eyeless gene
The region of the genome potentially containing the eyeless gene was isolated and amplified using a Polymerase Chain Reaction (PCR). In one eppendorf tube, the following were added in this respective order: 76.45mL ddH2O, 11mL 10X PCR Buffer, 6.6mL MgCl2(25mM), 2.2mL dNTPS (10mM each), 5.5mL of forward primer (10mM), 5.5mL of reverse primer (10mM), and 0.55mL of Taq Polymerase (Promega, USA). Out of this mix, 49mL was added to one PCR tube and 49mL was added to an alternative PCR tube. In one the tubes, 1mL of D. melanogaster DNA was added, and in the other, 1mL of H2O was added to serve as a control. Both tubes were placed in the thermocycler in which the following cycle was employed: heat start- 94o C for 2 min., melting- 94oC for 1 min, annhealing-41oC for 1 min., and elongation-72o C for 5 min. This cycle was repeated 18 times and held at 4oC.
PCR Clean-Up
A 1% agarose thin gel was prepared and the PCR products of the eyeless mutant gene were run next to wild-type genomic DNA. Both the eyeless band and wild-type band were identified and excised from the gel to be used in PCR clean up.
The PCR product was cleaned for all byproducts and nonspecific sequencing using the protocol described in the Wizard® SV Gel and PCR Clean-Up System kit (Promega, USA). The protocol was modified so that 75mL of membrane binding solution were initially used and 20mL of ddH2O was used for the step 12 resuspension.
End-A Cloning and Transformation
To add adenosine residues for cloning to the ends of the PCR cleaned-up product, the following reaction was set-up in a PCR tube: 1.0mL PCR Buffer, 0.2mL dNTP’s, 0.2mLTaq (Promega, USA) Polymerase, 8.6mL PCR product. The reaction was ran through the thermocycler under the following conditions: 94oC-2 min., 72oC-30 min., 4oC-hold.
The product of the PCR clean-up was ligated into a PGEM-T Easy expression vector using the protocol described in the pGEM®-T Easy Vector Systems Kit (Promega, USA). Three mL of PCR product was used for cloning. After a one-hour incubation, the sample was transformed into JM109 Competent Cells and spread on IPTG and X-gal LB plates and allowed to grow overnight at 37oC.
Miniprep of Plasmid DNA
Following bacterial growth, the isolated bacterial colonies were resuspended in LB + Ampicillin media before being Miniprepped according to the protocol in the Wizard® Plus SV DNA Purification System Kit (Promega, USA). Five mL of bacterial culture were used for this Miniprep. Once purified, the plasmid DNA was subjected to the following digest: 5mL plasmid DNA, 1mL 10x Buffer H, 1mL 10x BSA Buffer, 1mL Pst I. The digest was allowed to incubate @ 37oC for 1 hr. before the sample was run on a 1% agarose gel @ 100 mA for verification of the eyeless gene in the plasmid.
Sequencing of Plasmid DNA
The plasmids containing the eyeless mutant sequence as well as the wild-type DNA were sequenced using the Sanger Dideoxy-method according to the protocol outlined in the Thermo Sequenase® Labeled Primer Cycle Sequencing Protocol for USB® Kits (Li-Cor Biosciences, Lincoln, NE). Five hundred fmol of template was used for the reaction.
Analysis of DNA Sequencing
Both eyeless and wild-type sequences produced from the dideoxy sequencing were trimmed to remove plasmid sequence from analysis before being inserted into the BLAST database (8). Using the pGEM T-Easy sequence, it was determined where the eyeless gene was bound by plasmid, and any plasmid sequence was removed from analysis. The trimmed sequences were then entered into a nucleotide BLAST search under the following parameters: the “others” box was selected to include all databases and the nucleotide collection (nr/nt) was used (8). The BLAST results were used to identify which sequences contained the mutant eyeless gene. Because none of the proposed wild-type sequences showed significant identity, the Flybase database (9) was used to locate the entire eyeless wild-type gene, which was then placed with the confirmed mutant eyeless sequences in the “align two or more sequences” feature of BLAST (7). The alignment was then utilized to locate where the eyeless mutant demonstrated identity with the wild-type gene to more specifically locate the mutation.
Results:
It was hypothesized that a mutation to the eyeless gene in D. melanogaster flies would result in a phenotypic alteration and the mechanism of this mutation could be proposed upon further sequence analysis.
At the onset of experimentation, the observation of polytene chromosomes was employed to determine if the genetic mutation of eyeless was visible at a chromosomal level. It was found that while staining that verified the presence of DNA was perceptible, a sequence mutation could not be seen at the microscopic level. There was no observed difference between the wild-type and eyeless chromosomes.
The next technique utilized for genome analysis was PCR to isolate and amplify the portion of the genome containing the eyeless gene through the use of sequence-specific primers. After the PCR reaction, it was necessary to clean-up the product to account for non-specific binding of the primers as well as remove any byproducts that could form interference upon analysis. PCR had to first be confirmed on an agarose gel, and the confirmed bands were excised from this gel to be used in the gene clean-up (Figure 1). It was observed on the gel that the band for the mutant eyeless product (3,000 bp) shifted up in the gel when compared to the wild-type (2,000 bp). Using the DNA 1kb ladder for reference, this shift was shown to represent roughly 500 base pairs, indicating that the mutant eyeless gene contains a 500 base pair insertion compared to the wild-type.

Figure 1. Gel of PCR product demonstrating the shift of the eyeless mutant due to base pair insertion.
As is shown, the gel performed following the PCR clean-up showed that the eyeless mutant gene (Ey) had a band roughly 500 base pairs higher than the wild-type eyeless gene. The 1kb DNA ladder in the far left lane confirms this shift from the 3,000 and 2,500 base pair bands lining up with Ey and WT, respectively. This indicates that the mutant contains an insertion of 500 base pairs not present in the wild-type.
Once the PCR product was confirmed, it was ready to be ligated into an expression vector that would allow for stable and rapid growth while providing a means of the storage for the eyeless sequence. Before sequencing of the plasmid could be accomplished, it was necessary to confirm the presence of insert eyeless mutant sequence into the vector. This was done using a restriction enzyme digest, in which the sequence was cleaved from the plasmid before being run on a 1% agarose gel next to the wild-type sequence.
Once it was confirmed that the plasmid contained the desired genomic insert, the plasmids were ready for sequencing. The data extracted from the sequencing experiments produced a total of six different sequences: Seye1-T7, Seye1-Sp6, Seye2-T7, Seye2-Sp6, Swt1-T7, and Swt1-Sp6. T7 represented the product of the forward primer, while Sp6 stood for the reverse primer sequences. Upon trimming and BLAST analysis of these sequences, only Seye1-T7 and Seye1-Sp6 demonstrated identity with the D. melanogaster eyeless gene, as shown by their E values (Table 1). Greatest identity was seen with genomic eyeless sequence. The proposed wild type sequences lacked significant identity to D. melanogaster, but showed similarity to many generic expression vectors. This presented a problem for adequate comparison of the mutant to wild type genome. To account for this dilemma, a database of the entire D. melanogaster genome was utilized to provide the wild-type sequence (9).
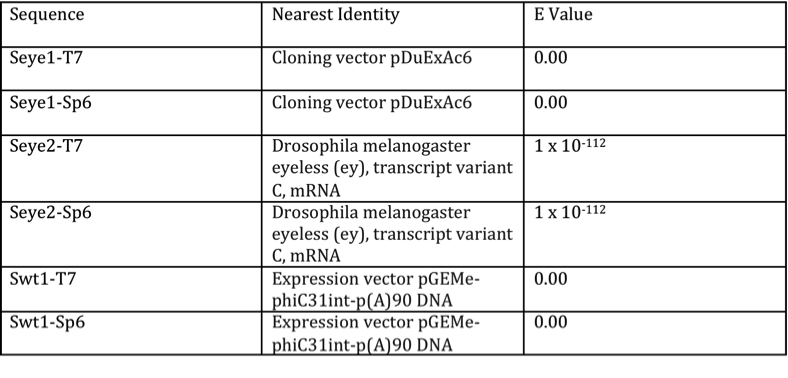
Table 1. Results of BLAST search for sequenced products
The chart demonstrates the identities and E values found for the BLAST of the sequenced products. Seye1-Sp6 and Seye2-T7 showed identity to eyeless of D. melanogaster, verifying them as the mutant sequence. The remaining sequences showed identity with expression vectors.
The entire D. melanogaster wild-type eyeless sequence, including introns, was used for a second BLAST search, this time aligning the mutant eyeless sequence with the FlyBase wild-type. It was found that beginning in exon 2, at the 7,528th nucleotide, from the beginning of the eyeless wild-type gene, the product of the forward primer begins. Identity continued through intron 2 to the 8,262nd nucleotide. With the reverse primer product, identity was noted beginning at the 9,165th nucleotide of exon 3, continuing into intron 2. What was discovered was that the forward primer showed near identity and the reverse primer demonstrated significant identity, indicating that the mutation must exist in the sequence region in-between, not bound by the primers. This would place the proposed mutation sequence within intron 2 (Figure 2).
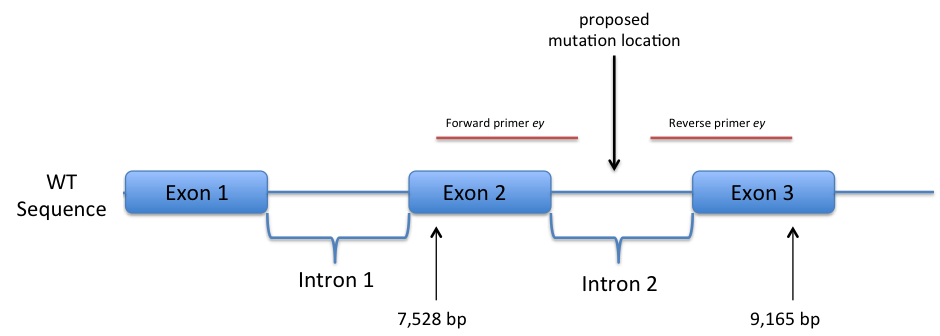
Figure 2. Side-by-side comparison of wild-type and mutant eyeless sequences following first round of sequencing. (not to scale)
As is shown, the eyeless gene sequenced from the plasmid, when compared to the wild-type D. melanogaster genome, showed identity beginning in exon 2, through intron 2, and ending in exon 3. The entire eyeless gene sequence within intron 2 was not sequenced due to the primers’ limited replicative abilities and the size of the gene. Because significant identity was demonstrated for both the sequence from the forward primer and the sequence from the reverse primer, respectively, the mutation that would include variation from wild-type sequence must lie within intron 2.
Identification of the location of the variant nucleotides within the mutant eyeless gene allowed for further hypothesis of the mutational mechanism.
Discussion:
The given experiment provides a means for not only distinguishing the genomic identity of a mutated eyeless gene of D. melanogaster from its wild-type equivalent, but demonstrates the necessary course of study for narrowing in on a mutation at the DNA sequence level. More so, the sequences obtained allow for further analysis and research consideration given their ability to place the mutation within a specific chromosomal region.
It was demonstrated that examination of the polytene chromosomes from both the wild-type and mutant flies was insufficient for determining substantial variance that could be attributed to mutation. This promoted the use of DNA sequencing for analysis.
Once the genomic sequence was properly extracted and prepped for sequencing, the sequences generated could be properly analyzed to provide reasonable mutational explanation. It was found that the mutant eyeless gene begins in exon 2, extends through intron 2, and ends within exon 3, relative to the wild-type eyeless gene. In the identity with wild-type shown from the forward and reverse primer products of the mutant sequence, there was little variation seen. This means the mutation must lie in the region of the eyeless gene not sequenced by the initial priming round, being intron 2. This mutational placement promotes three possible mechanisms for change in phenotype.
It must be recognized that any explanation from this point forward must fit the model of the mutation existing within an intron. One hypothetical explanation would be a mutation in the scaffold attachment region (SAR) of the chromosome. SAR’s are conserved DNA sequences within introns that aid in attaching chromatin of eukaryotic chromosomes to the nuclear matrix, providing necessary stability for DNA replication (10). Additionally, the conserved sequences provide stability during transcription, not by being transcriptionally active, but by insulating the process (11). A mutation preventing nuclear matrix attachment would disrupt this stability, and it is likely that replication or transcription would not occur (11). Because SAR’s exist in introns, it is feasible that the eyeless mutant gene sequence mutation would be disrupting an SAR sequence. What refutes this reality is the existence of phenotypic change within the eyeless mutant flies. In that a mutation within SAR sequence prevents replication as well as transcription, such a mutation would result in no expression of that gene whatsoever. If this was the case, no phenotypic effect would be expected, only a lack of gene expression altogether. As was previously indicated, the phenotype for the mutation of eyeless seems to be variable expressive, not a complete lack of gene expression.
A second consideration for a mutation within an intron would undoubtedly involve splicing mechanisms. There are several factors that make this suggestion unlikely. In that the sequences obtained from the dideoxy sequencing include both borders of the exons and intron 2, the conserved sequence within mRNA that is necessary for proper splicing appears to be conserved and unmutated. Additionally, if the mutation, instead, affected spliceosome machinery, the resulting transcriptional and translational manifestations would be a global cellular issue in that no mRNA within the cell would be able to be spliced properly. This is not what is phenotypically observed in the eyeless mutant flies. It is, therefore, unlikely that the mechanism of mutation works through the splicing mechanisms.
A final, more sufficient proposition for an intron mutation includes gene expression amount and the enhancement or repression of gene expression, transcriptionally. Because enhancers and repressors must bind to conserved sequences on the DNA undergoing transcription, it is valid to consider that one such sequence could exist within the intron of the eyeless gene of D. melanogaster. If this enhancer or repressor sequence were mutated, expression levels of the eyeless gene would be altered.
In order to further validate the hypothesis that the mutation within the eyeless gene involves an enhancer or repressor sequence, it would first be necessary to complete sequencing of the entire eyeless gene through intron 2. This could be done in the same manner as before, using the Sanger Dideoxy-method, with new primers designed for roughly every 800 base pairs. Analysis would need to be performed again in the same way as before with utilization of the BLAST database and entire D. melanogaster eyeless wild-type gene for comparison. Finally, it would be beneficial to monitor gene expression in wild type flies and mutant flies to observe variation in expression quantity. This could be achieved through protein assays performed with proteins resulting from eyeless expression. For this to be achieved, it would first be necessary to identify all proteins involved in the signal transduction pathway promoted by eyeless expression. Additionally, both the wild-type eyeless and the mutant eyeless gene could be applied to a DNA microarray using a genome chip (12). This would allow amount of gene expression to be monitored to see if a difference exists. If the mutation were affecting a enhancer or repressor sequence of the eyeless gene, it would be expected that significant variation in gene expression would be observed. Such research would permit further definition of the eyeless mutation in D. melanogaster flies.
References:
(1) Understanding Evolution. 2004. University of California Museum of Paleontology.
grant nos. 0096613, 0841757, and 0918741.
(2) Morgan, T.H. 1910. Sex-limited inheritance in Drosophila, Science, 32:120-122.
(3) Ashburner, M. 2012. Drosophila melanogaster: Common Fruit Fly. Encyclopedia
- of Life.
(4) Arnini, C. 2012. Using Drosophila to Teach Genetics. Yale-New Haven Teachers
Institute. Unit 96.05.01.
(5) Otto, P.A. 2000. Drosophila Mutant Phenotypes. Genetics and Molecular Biology:
CGS. (23)4.
(6)Focusing on the eyeless Gene. SCIENCE · VOL. 267 · 24 MARCH 1995 pp. 1766-1767.
(7)Culture of Drosophila: The Laboratory Setup. Cold Spring Harb Protoc; 2007;
doi:10.1101/pdb.ip34.
(8) Basic Local Alignment Search Tool. National Library of Medicine.
(9) A Database of Drosophila Genes and Genomes. 2012. Flybase. Version
FB2012_06, released November 6th, 2012.
(10) Razin, Sergey. 2005. Matrix‐associated Regions (MARs) and Scaffold
Attachment Regions (SARs). Russian Academy of Sciences. DOI:
10.1038/npg.els.0005035.
(11) Bode, J., & Gluch, A. 2008. Scaffold/matrix attachment regions (S/MARs):
relevance for disease and therapy. Helmholtz-Zentrum für Infektionsforschung MBIO/Epigenetic Regulation, Inhoffenstrasse 7, Braunschweig, Germany. DOI:10.1007/978-3-540-72843-6_4.
(12) Shi, L. 1998-2002. DNA Microarray (Genome Chip) — Monitoring the Genome on
a Chip. www.Gene-Chips.com.
