Structural Elucidation and Subsequent Compound Derivation via the Direct Observation of Chemical, Physical, and Spectroscopic Molecular Properties
A Qualitative Analysis of Two Organic Unknowns:
Structural Elucidation and Subsequent Compound
Derivation via the Direct Observation of
Chemical, Physical, and Spectroscopic Molecular Properties
Introduction
The characterization and classification of an unknown organic compound (or multiple compounds) is a frequently implemented lab testing the cumulative skills of undergraduate students or lab novices at various Universities and related institutions. Moreover, necessitating students to apply various techniques learned throughout the lab course exhibiting individually varying degrees of retention in us of previously comprehended techniques. This illustrates that analysis of an unknown molecule in certain scenarios, which one lab TA described as “real-world chemistry”, is not as simple or clear-cut as entry-level chemistry textbooks often suggest. Critical thinking and problem-solving are prerequisites to successfully classifying unknowns. Accordingly, compounds should be treated as if they are toxic initially, as there were potentially toxic compounds that could be assigned from the Sigma Aldrich chemical company product catalogue; the Sigma Aldrich Product catalogue contains somewhere around 5,000-7,000 compounds. Accordingly, the empirical determination of melting point or boiling point as well as compound solubility in organic solvents is a significant first step in analyzing an unknown. The next step, is interpretation of what the physical properties (solubility and the melting or boiling point temperatures)
CFH Allen contributed to the popularization of 2,4-dinitrophenylhydrazine in a 1930 review article which employed this reagent to determine practicality of it as a functional group test for the presence of carbonyl.1 He postulated that earlier chemists must not have found it useful since they were most likely only applying this reagent to aromatic compound, which were already known to have formed crystalline products with phenylhydrazine.1 Allen(1) and lab technicians/students in the chemical laboratory of McGill University(1), conducted the earliest known, and most diverse research utilizing a total of 26 different Aldehydes and 33 Ketones, in addition to 8 “Kjeldhal” carbonyl-containing compounds.1 Interestingly, one of the cyclic aliphatic ketones mentioned within this article also happened to be what I have determined as unknown sample L2-11, cyclohexanone. The 2,4-dinitrophenylhydrazine derivative of cyclohexanone in Allen’s(1) article is found within Table 1 and has a recorded melting point of 160oC and a orange-yellow color.1 In summary, Allen determined this to be an effective and inexpensive reagent useful in the preparation of crystalline carbonyl derivatives of aliphatic compounds as well certain cyclic compounds (but indicated that precipitate formation was not as general).1 Conversely, this reagent is not suitable to employ with α-hydroxyketones due to the difficulty in separating the mixtures formed.1
2,4-Dinitrophenylhydrazine can be prepared from a benzene starting material by first reacting the benzene with Cl2/FeCl3 to achieve the formation of chlorobenzene. Then, two equivalents of nitronium ion will undergo electrophilic aromatic addition to the monosubsitituted chlorobenzene.1 The nitronium ions formation requires prior acidic workup in a redox reaction between sulfuric acid (reduced) and nitric acid (oxidized). The final reaction of the trisubstiituted benzene involves the addition of potassium acetate and hydrazine sulfate, giving rise to the desired reagent 2,4-dinitrophenylhydrazine.1
Unknown Liquid L2-11
Unknown liquid L2-11 has been determined with near certainty to be cyclohexanone based both on the spectral signals of the species and on the physical and chemical data collected throughout the unknown lab. However, determination of this compound was not simple due to the highly reactive nature of the alpha-beta-gamma saturated cyclic ketone. This leaves 2 chemically different, acidic, unhindered proton types susceptible to extrapolation from electrophilic species (referring to the 2 and 6 locations; the 4 total alpha protons (the most acidic) and the protons bound to the 4th carbon, the gamma carbon, the other area in which electron delocalization results in a partially negative charge increasing the acidity of its protons).
The unknown organic liquid assigned to me on April 23, 2013, consisted of a slightly cloudy but otherwise transparent and clear solution. It smelled strongly of alcohol which eliminated the need to hold the vial near my face to smell the unknown (avoiding inhaling any potentially toxic fumes). The micro boiling point analysis was the first test performed (after separation of half of the liquid into another vial, of course) in a silicone-based solvent which was not volatile or reactive and had a high boiling point. This allowed for precision in boiling point analysis as the temperature observed was consistent with the boiling point provided in the sigma Aldrich MSDS as well as in the text book and other scholarly sources.1
The procedure to determine boiling point involved the addition of roughly one mL of liquid unknown to a test tube followed with placing a microcapillary tubule (with its convex surface oriented toward the surface) into the test tube with the liquid. The test tube was anchored securely to a thermometer with a tightened rubber-band. A silica mixture was heated due to its unreactive nature allowing for the elimination of any hot-water bath solvent type limitations to be taken into account. The boiling initiated once the microcapillary tubule transitioned from releasing oxygen bubbles into the lab tube to sucking in the oxygen bubbles. This point was recorded twice, and was determined to begin bubbling around 154oC the first time and 155oC the second time. The heat source (hot plate) was then removed and the point at which the inverse of the prior transition occurred was recorded at about 157oC the first time and 156oC the second time. This analysis was performed twice with a (+/-) 1o C or less margin of error, leading to a conclusion that the laboratory boiling of my unknown was ~156oC.
The solubility of my liquid unknown was not apparent in any of the solvents tested for; water, sodium hydroxide, hydrogen chloride, and sulfuric acid did not change the small amount of my liquid as far as optical observations go, and there was no thermodynamic response such as a localized increase of kinetic energy. Therefore the unknown was classified as an inert compound but I didn’t place too high of a degree of emphasis on this analysis since the 5% concentration of the solvents used to test solubility are not exactly the most accurate indicators. The solubility of my solid followed suite; it proved to be entirely insoluble in all of the solvents listed above. The last analysis employed for the liquid unknown on the first day of the lab was to collect the Infrared Spectrum which would reveal potentially testable functional groups for the following lab. The liquid IR had a very strong sharp peak in the 1720-1710cm-1 range, which is indicative of an α,β-saturated ketone (i.e., unconjugated). It is also noted that the aliphatic C-H stretches around the 2,950-2,800cm-1 region were characteristically sharp and strong, and the manifestation of these peaks provided insight
The following lab period (4/25/13), molecular saturation was tested in the liquid unknown. This test entailed adding a reaction mixture comprised of methylene chloride (1 mL) and 4% bromine (l) (3/4 mL) in a drop-wise manner to the liquid unknown. If the unknown mixture remained the same color as it was before addition of any Bromine/CH2Cl2, then subsequent drops were added in a droprwise manner up to ~5 drops.If no change was yet observed, it indicated that the molecule was either saturated or that a substitution reaction was occurring. Due to the absence of any optical or thermal reaction in my product after the first five drops, I suspected that a substitution reaction was most likely occurring due to the absence of any characteristically conjugated peaks on the IR spectrum to the left of 3,000 coupled with the present results.
The test which reaffirmed my postulation derived from the IR spectra pertaining to an aliphatic molecule was the addition of 2,4-dinitrophenylhydrazine to my liquid unknown.1 The addition of only one drop of 2,4-dinitrophenylhydrazine resulted in the rapid formation of a darker yellow precipitate. The presence of precipitate, however, is only indicative of the presence or absence of a carbonyl group. The color of the precipitate formed and the consideration of reactivity rate differentiates an aliphatic compound from an unsaturated compound. Benzylic compounds form a dark orange/red precipitate whereas fully saturated (unconjugated) ketones (excluding methyl-hydroxy ketones) form much brighter yellow precipitate.1
The Tollen’s test (2Ag(NH3)2OH) resulted in a positive result for my liquid unknown, owing to the reactivity and high acidity of the alpha protons adjacent to the saturated cyclic carbonyl group. It was noted throughout the literature that Tollen’s test could provide a false positive for methyl ketones (correspondingly the high pKa values of my alpha protons are just as reactive if not more reactive than methyl ketones due to the additional factor of ring strain in cyclohexanone, while not very great, still exists). The data extrapolated from this test regarding the physical properties of my unknown threw the evidence I had collected throughout this procedure into question; this is precisely why qualitative analysis is so important, because going through the frustrating process of elimination followed by re-evaluation due to unpredicted results further emphasizes the unpredictability associated with chemistry outside of a textbook. I was expecting a 1H NMR signal to appear downfield (9-10 ppm) in the aldehyde region after this test. However, one did not appear which resulted in further confusion, only to be alleviated after determining the splitting pattern indications and collecting reference 1H NMR spectra from the spectral database. The database spectra for cyclohexanone matched very closely to my unknown structure; the reason for seeking the spectra of cyclohexanone to begin with is because I had postulated it to be the unknown liquid compound due to the absence of a methyl group, indicating a cyclic ketone. With this information taken into account, as well as all of the previous information, it was reasonably determined that organic unknown liquid L2-11 is cyclohexanone, which is has a boiling point in the Sigma Aldrich MSDS of 155oC.
The lab following the acquisition of the 1H NMR spectra for both the solid and liquid (5/2/13), the cyclohexanone derivative was formed using 2,4-dinitrophenylhydrazine once more to form the derivative. However, this time I followed the instructions highlighted in the appendix in the back of our lab textbook for derivative formation procedures. The instantaneously formed derivative precipitate was filtered and washed under vacuum filtration in a Bucchner funnel. The Melting point of crude 2,4-DNP cyclohexanone derivative was only ~2 degrees lower than that of the pure form, indicative of the relatively instantaneous purity obtained from this reaction.1
Solid S2-11
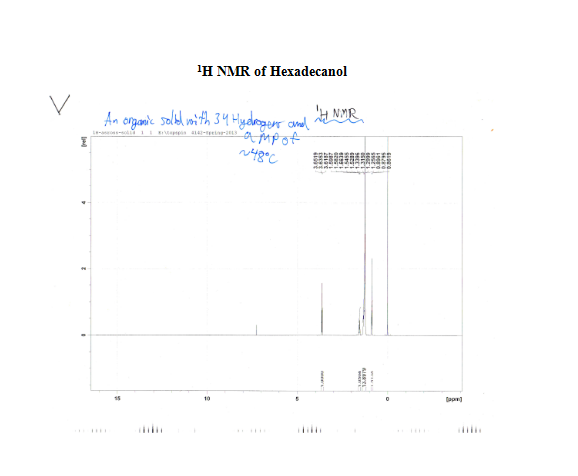
The solid unknown obtained on 4/23/13 proved to be more of a challenge as far as structural determination and physical properties are concerned relative to my liquid unknown. The melting point collected on the first day of lab was about 47-48oC.2The solid was entirely insoluble in all of the previously mentioned solvents (HCl, NaOH, H2SO4, and water) in any of the solvents with a 5% concentration used to determine molecular physically properties was not evident; rather, it appeared to be entirely insoluble.
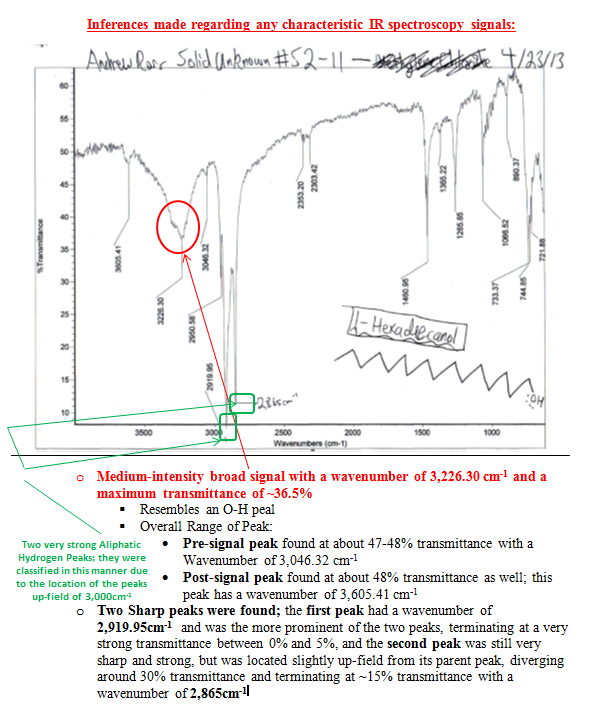
The spectra are discussed in the appendix in more detail, but it was clear that my solid was an alcohol due to the diagnostic broad and strong signal in the 3000-3500 cm-1 region and the lack of a strong or sharp peak in the region of 1550 to 1750cm-1, which would denote the presence of a carbonyl group C=O. The three pieces of information from the first day of lab provided useful insight into what my product could be.
The saturation test (performed at the start of the second lab on 4/27/13) was achieved by adding a reaction mixture comprised of methylene chloride (1 mL) and 4% bromine (l) (3/4 mL) in a drop-wise manner to the solid. The solid was dissolved in tetrahydrofuran (THF) since it was insoluble in water. The solid provided an instantaneous negative test result, with the red bromine color forming immediately after the addition of only one drop indicating that the solid unknown was a saturated hydroxyl (as long as the IR spectra functional group indication holds true).
The 2,4-dinitrophenylhydrazine test was also performed on the solid for the sake of experimental precision, just in case of a hidden C=O bond on the IR and the presence of a carboxylic acid. As expected, there was no result and this negative result helped provide additional evidence that it was an alcohol.
After the saturation test an attempt to run the Lucas test on the unknown solid compound was made even with the warning in the text regarding this test as a poor test for a solid alcohol; however, the rational made at the time was that the unknown solid at hand had a melting point barely 20oC higher than room temperature, therefore it might react to a higher degree. Lucas Reagent (ZnCl2) and hydrochloric acid are added to the dilute solid (dissolved in 1 mL of THF), and the alcohol is seen to dissolve if it is primary at room temperature, whereas 2o alcohols give slight cloudiness and 3o alcohols give moderate cloudiness. Since nothing was observed, in fact nothing even dissolved, it was determined that if my compound was an alcohol it was both a primary alcohol and an alcohol greater than six carbons in chain length, due to its insolubility in the Lucas reagent.
To determine if my solid might have an amine functional group, I tested for the presence of one. 0.05g of my unknown solid was dissolved in tetrahydrofuran, to which acetonitrile was added in a first experiment due to not having access to acetochloride as the text book suggests. However, upon reconsidering the nucleophilic properties of nitrile, I decided that another reagent might be more similar to acetochloride as far as nucleophilicity and acidity are concerned. Therefore, I decided to try this procedure again but this time I employed benzoyl chloride as my reagent. There was no reaction once again, diminishing the possibility of an NH2 functional group.
The last test I ran before being able to record my 13C NMR was the Chromic acid test since nothing else seemed to work with my unknown (at this point thought to be either 1- or 2-hexadecanol based on the 1H NMR already obtained, of the two I was 90% sure of the former) compound and this was carried out by dissolving 0.01g of unknown solid in 1mL of reagent grade acetone. This procedure was first tested on ethyl alcohol to ensure that the anticipated reaction would occur being the formation of a blue-green color within 2 seconds of the addition of Chromic acid. The control reaction went exactly as I had anticipated; however, the reaction I anticipated my solid unknown to undergo did not go exactly as planned because while it did slightly change colors, it did not appear to me as a blue or green color, or a further intensification of the original orange color for that matter. However, being that this is the only line of evidence I have to go off of for any functional group tests for my unknown solid, this provides the best support that my solid is a primary alcohol being that it actually reacted in a reaction mixture rather than remaining insoluble throughout the course of one.
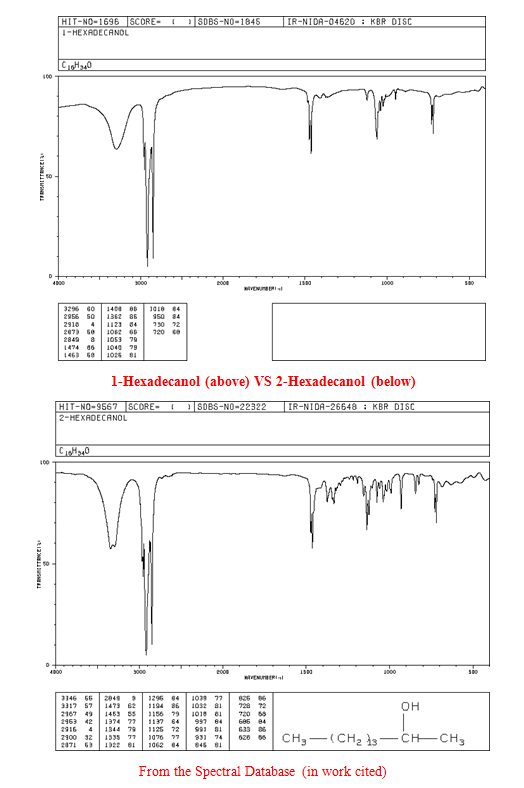
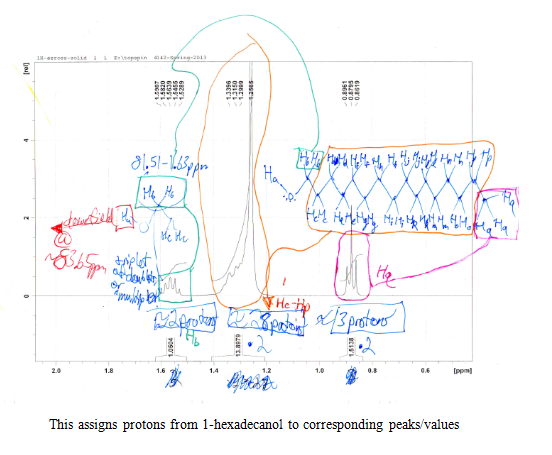
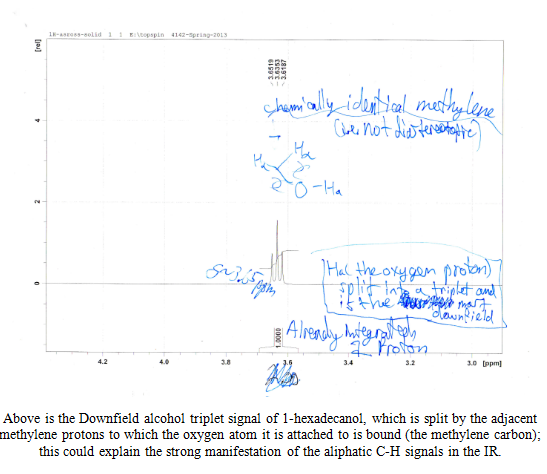
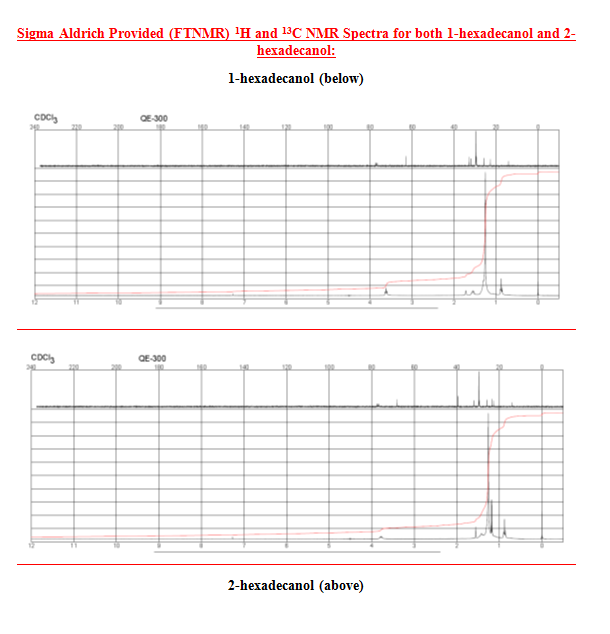
After the collection of my 13C NMR it was made very clear that I had 1-hexadecanol based on the discrepancy in terminal downfield peak position between the two. The 13C NMR revealed the signal to be at 63.168ppm, which is very close to the predicted value on the Sigma Aldrich product guide for 1-hexadecanol, much further than the 68-69ppm signal predicted for 2-hexadecanol. The additional outlier is in 2-hexadecanol around 39ppm, whereas 1-hexadecanol has the second peak clustered near the rest of the carbon peaks at 32 ppm. This is because of a greater number of chemically different carbons within 2-hexadecanol, and reflects the increased electron delocalization disparity by having slightly further downfield peaks coupled with a slightly lower melting point.
The derivative of 1-hexadecanol was formed the following lab period, starting with dissolving 0.25g of 1-hexadecanol in 1.5mL of pyridine and adding 0.25g of 3,5-dinitrobenzoyl chloride. Next the mixture was heated under reflux for 15 minutes and then was cooled and and poured into a cold reaction mixture of 2.5mL water and 2.5mL 5% sodium carbonate. This was continuously stirred through the use of a magnetic stir bar until the temperature became low enough to the point that crystallization was feasible. After crystallization in an ice bath, it was collected in on a Bucchner funnel and the product was filtered via vacuum filtration and cold water washes. This product was then recrystallized using 100% ethanol heated to dissolve the crystals and allowed to crystallized, then after addition of the crystals to the bucchner funnel cold H2O was added as a wash for further purification.
There are two major reasons why my product results are skewed. The first reason being, according to Robinson et al.2 high weight carbon molecules do not proceed with phthalization after undergoing acetylation, because it is not soluble enough in pyridine.2 This means that essentially my reaction was set up for failure by not taking enough cautious steps beforehand. The fact that the melting point of my unknown is 49oC (in literature, 48oC in lab) and the method chosen to form a 3,5-dinitrobenzoate derivative in the present report from 1-hexadecanol had inherent flaws. By using the procedure given for solid alcohols, I failed to notice that the melting point of 3,5-dinitrobenzoyl chloride is higher than that of my alcohol solid, and upon the addition of heat, as occurs in the liquid alcohol procedures section, my alcohol would have had a much higher probability of forming a pure derivative.
Overall, the results and experience gained from the unknown lab have helped develop an increased familiarity with why each step is structured as it is. In retrospect, the methodology employed in derivative formation from my solid would have been shifted to the liquid procedure rather than using the solid procedure. That would increase the statistical probability of a pure derivative forming since pyridine would have been removed from the reaction.
References Cited:
(1) THE IDENTIFICATION OF CARBONYL COMPOUNDS BY USE OF 2,4-DINITROPHENYLHYDRAZINE. Charles, C.F.H.; Journal of the American Chemical Society. 1930, 52(7), 2955-2959.
(2) Rapid Determination of Organic Hydroxyl Groups with 3,5-Dinitrobenzoyl Chloride. Robinson, W.T., Jr.; Cundiff, R.H.; Markunas, P.C. Analytical Chemistry. 1961, 33(8), 1030-1034.
Physical Data Sources and Spectral Sources for Unknowns (attached to the report as well)
Cyclohexanone
– Chemical Properties
– 1H NMR
– IR
– Derivative IR
– Sigma Aldrich
1-hexadecanol (product of solid S2-11)
- Chemical Properties
- Sigma Aldrich
- 1H and 13C NMR
– MSDS
2-Hexadecanol (chemically related compound with similar properties; not my unknown)
- 13C NMR
Appendix
The Solid Unknown S2-11: 1-Hexadecanol
– Color—White
– Odor—Very slight scent of something stale; however, the aroma is not pungent and is not nauseating or irritating
– Appearance—Crystalline (white) solid with some larger crystals stuck together; seems crude (somewhat), does not resemble the purified fine crystals collected earlier in the semester from recrystallized samples.
– MP—46-48oC—low Melting Point, aided in compound determination