Formation of Carbon-Carbon Double Bonds via the Wittig Experiment and the Hydrolysis of the Ester
Written by Dan
Abstract
In this experiment, a double bond was formed between two carbon atoms following the Wittig mechanism (discovered in 1954 by George Wittig[1]) to see if the resultant carboxylic acid product mixture is made up of a single diastereoisomer, or a mixture of the two. It was found that only one diastereoisomer was formed.
Introduction
|
Figure 1 – reaction scheme for the overall reaction
|
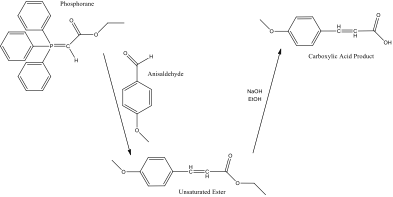
The reagents required for the experiment were an ylide (a species with a positive and negative charge on adjacent atoms, in this case a phosphorane) which reacts with a carbonyl (in this case, anisaldehyde) to give an unsaturated ester. This ester is then hydrolysed in basic conditions to give a carboxylic acid (figure 1).
|
Figure 2 – reaction mechanism of the Wittig reaction
|
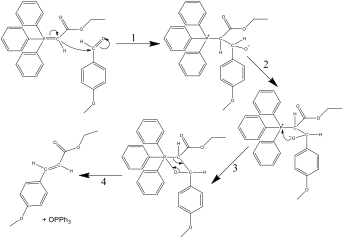
To obtain the phosphorane, the corresponding phosphonium salt must be reacted with sodium hydroxide. The mechanism for the reaction of the phosphorane and anisaldehyde is shown in figure 2[2]:
In step 2, there is rotation about the bridging carbon-carbon bond, which means that the oxygen anion can be close enough to the phosphorus cation to react, to form Ph3PO and the unsaturated ester. Because the molecule can only be in this orientation for the oxygen to react with the phosphorus, only one of the two possible geometric isomers of the unsaturated ester is formed, and the reaction is either E or Z selective, dependant on the structure of the ylide. Stabilised ylides (those that can further conjugate away from the P=C bond) tend to mainly from E isomers as their products[3]. This suggests that on TLC analysis of reaction progress, there will only be one product spot. The name of the carboxylic acid formed was found to be p-methoxycinnamic acid[4].
Experimental
a) Preparation of the phosphorane
A Phosphonium salt (25.48g) was dissolved in deionised water (300cm3) in a 1L conical flask. Some solid remained, so more water (100cm3) was added. Any undissolved or discoloured solids were removed by gravity filtration. The mixture was cooled to -5°C in an ice bath, after which sodium hydroxide (34cm3, 2M) was added to the mixture. A sticky white solid was produced, which then was dissolved in dichloromethane (DCM) (100cm3). The phosphorane was then extracted from the aqueous layer using a separating funnel, and two further washes of DCM (2 x 50cm3). The organic layers were then combined and dried with anhydrous magnesium sulphate. This was filtered under gravity and the solvent was removed under reduced pressure to produce an oily residue, which then crystallised.
b) Reaction of the phosphorane with anisaldehyde
The phosphorane was redissolved in DCM, and added to anisaldehyde (8.4cm3) in DCM (50cm3) over about ten minutes. The mixture was then brought to reflux, and the colourless solution gained a yellow tinge. Reaction progress was monitored by TLC (developed in a 4:1 mixture of 40/60 petroleum ether: ethyl acetate, and visualised under UV light). Once the reaction was complete, the solution was left to cool, and then the solvent was removed under removed pressure. 40/60 Petroleum ether (200cm3) was added to the resulting yellowish oil, and left to stir for around 30 minutes. A white powder was formed, and was filtered at the pump using a coarse sinter funnel. A further two portions of 40/60 petroleum ether was used to wash the solid (2 x 30cm3). The solvent was removed under reduced pressure to produce an oil (the unsaturated ester). Crystals formed with agitation, and 60mg of this solid was kept as a reference.
c) Hydrolysis of the unsaturated ester
The crude unsaturated ester (1.53g) was added to a 3:1 mixture of 10% sodium hydroxide: ethanol (20cm3). The resulting solution was brought to reflux. Progress of the reaction was monitored by TLC analysis (developed in DCM, visualised under UV light). During the reflux the solution turned a deep yellow colour. Once the reaction was complete, the product was extracted with water (10cm3) and the solution was allowed to cool to room temperature. Any solids that formed at this stage were filtered under gravity, to produce an egg yellow coloured solution. The solution was acidified to pH2 with concentrated hydrochloric acid to form an egg yellow solid. The resultant mixture was filtered at the pump and washed with ice water (2 x 25cm3) followed by a 3:1 mix of cold water: ethanol (10cm3) to isolate the yellow solid, the crude carboxylic acid (p-methoxycinnamic acid) (2.09g, 158%). This product was recrystallised to give a more pure sample of the p-methoxycinnamic acid (0.10g, 7.56%; mp 180°C (ethanol); vmax/cm-1 (nujol mull, NaCl plates) 3394.5 (O-H), 2953.86 (C-H), 1930.7 (aromatic overtones), 1459.2 (C=O), 1380 (C-O); δH (400MHz; DMSO) 12.2 (1H, s, CO2H), 7.65 (2H, d, J 8.78, H4, H5), 7.55 (1H, d, J 15.97, CH=CHCO2H), 6.98 (2H, d, J 8.77, H6, H7), 6.39 (1H, d, J 15.98, PhCH=CH), 3.8 (3H, s, PhOCH3); δC (100MHz; DMSO) 168.73 (CO2H), 161.79 (CB), 144.61 (CH=CHCO2H), 130.79 (CC), 127.67 (CE), 117.34 (Ph-CH=CH), 115.18 (CD), 56.12 (PhOCH3))
|
Figure 3 – proton and carbon labelling scheme
|
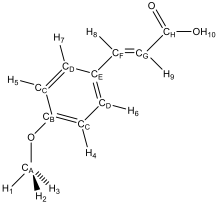
Where a proton has been assigned a subscript number, and a carbon has been assigned a subscript letter, it is in accordance with the naming scheme given in figure 3.
Results
For TLC analysis of the reaction between the phosphorane and anisaldehyde, and the hydrolysis of the unsaturated ester, refer to the laboratory notebook (pages 14 and 15). Details of the masses and yields of the crude ester, and both crude and recrystallised p-methoxycinnamic acid obtained can also be found in the laboratory notebook. All information in the laboratory notebook has been marked previous to the writing of this report.
An infra-red spectrum of the recrystallised p-methoxycinnamic acid is included in the appendices (appendix 1), and information about the peaks in the spectrum is tabulated below:
|
Wave number / cm-1 |
Assignment of band: Functional group |
|
3394.5 |
O-H (str) |
|
2953.86 |
C-H (str) |
|
2924.67 |
C-H (str) (Nujol) |
|
1930.7 |
Aromatic overtones |
|
1600 |
C=O (str) |
|
1459.2 |
C-H (bend) (Nujol) |
|
1380 |
C-O (str) |
Refer to the appendices for copies of the provided 1H NRM spectra of the start materials (anisaldehyde, appendix 2) (phosphonium salt, appendix 3), and for proton assignment for these spectra, refer to appendix 4.
A 1H NMR spectrum of the p-methoxycinnamic acid is included in the appendices (appendix 5). Information about the peaks in the spectra has been tabulated below.
|
Chemical Shift (δ) / ppm |
Multiplicity |
Coupling Constant (J) / Hz |
Integral (distance measured) / mm |
Assignment (proton number) |
|
12.2 |
Singlet |
– |
– |
10 |
|
7.65 |
Doublet |
8.78 |
40 |
4, 5 |
|
7.55 |
Doublet |
15.97 |
20 |
9 |
|
6.98 |
Doublet |
8.77 |
39 |
6, 7 |
|
6.39 |
Doublet |
15.98 |
19 |
8 |
|
3.8 |
Singlet |
– |
60 |
1, 2, 3 |
A 13C NMR of p-methoxycinnamic acid has been provided and a copy of it can be found in the appendices (appendix 6). Assignment of the peaks can also be found in the appendices (appendix 7).
Where a proton has been assigned a subscript number, and a carbon has been assigned a subscript letter, it is in accordance with the naming scheme given in figure 3.
Melting point analysis of the recrystallised hydrolysis product gave a melting point range of 180-181°C
Discussion
The documented melting point for the p-methoxycinnamic acid is 173-175°C[5]. This suggests that the sample of product obtained was not entirely pure. This may be due to contaminations in the sample, such as solvent that had not been removed completely (water, DCM, 40/60 petroleum ether) or unreacted compounds (sodium hydroxide, ethanol, hydrochloric acid, anisaldehyde, phosphorane).
The 1H NMR spectrum of the recrystallised product (appendix 5) shows possible evidence of impurities with low intensity peaks at 1.25ppm and 3.35ppm. However, product peaks did occur in the predicted 1:1:1:2:2:3 ratio suggesting that the reaction had proceeded as planned, and that p-methoxycinnamic acid had been synthesised.
In the introduction, it was stated that stabilised ylides (the phosphorane used in this reaction was a stabilised ylide) tend to form E- isomeric alkenes after reacting with carbonyls via the Wittig mechanism. We can see that this was the result of the reaction carried out by inspecting the 1H NMR (appendix 5) of the recrystallised product. Figure 4 shows the orbital positions in C-H bonds in E- and Z- geometries of a C=C bond and compares them to the positions of the orbitals of C-H bonds in a benzene ring.
|
Figure 4 – comparison of orbital positions in E- and Z- isomers with a benzene ring
|
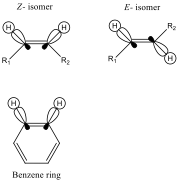
We see that the positions of C-H bonding orbitals in the Z- isomer are exactly parallel (this is fixed due to the lack of rotation about the C=C bond), and are very similar to those of the C-H bonding orbitals in a benzene ring. This gives a good overlap and allows for a good level of ‘communication’ between the two protons, and gives a moderate coupling constant, J, value when splitting each other. We see that in the E- isomer, the C-H bonding orbitals are orientated exactly antiparallel to each other. As predicted by the Karplus relationship[6], when the dihedral angle between hydrogens (and orbitals) is 180° (as in the E- isomer), the coupling constant, J, is larger than when the dihedral angle between hydrogens (and orbitals) is 0° (as in the Z- isomer and benzene).
If the hydrogens connected to the C=C bond of p-methoxycinnamic acid were cis- to each other, it would be expected that the coupling constant, J, due to H9 observed in the peak due to H8 (and vice versa, figure 3) would be similar to the coupling constants that arise due to H4 and H5 in the peaks due to H6 and H7 respectively (and vice versa, figure 3).
|
Figure 5 – a demonstration of the E- selectivity of the mechanism with a stabilised ylide
|
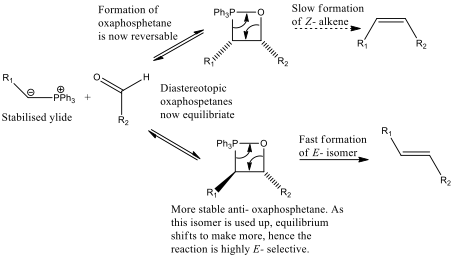
What is actually observed is a much larger coupling constant (almost 200%) in the signals from H8 and H9, indicating that they are trans- to each other and that the E- isomer was formed. It is generally assumed that the E- selectivity of the Wittig reaction with stabilised ylides assumes that the formation of the oxaphosphetane, steps 1 and 2 in the mechanism (figure 2), is reversible. If this is the case then the reaction is thermodynamically controlled. Providing that the elimination of Ph3PO is slow compared to the interconversion, the stereospecific step no longer reflects the initial kinetic ratio of oxaphosphetane diastereoisomers. It is not wild to assume that the more thermodynamically stable diastereoisomer is the isomer with the bulky groups as far away from each other as possible (trans- to each other) to avoid steric clash. This relationship is demonstrated in figure 5[7].
At 0.1g, 7.56%, the yield of the reaction was extremely low, especially since 2.09g of product was isolated after rotary evaporation of the solvent. This may be due to the fact that crude ester was hydrolysed. If the crude ester had been recrystallised before it was hydrolysed, the yield may have been a lot higher. If the experiment were to be repeated, it would be interesting to see if the yield of p-methoxycinnamic acid improved when hydrolysing a recrystallised sample of ester rather than the crude ester.
Conclusion
The hydrolysis of the unsaturated ester formed as a result of the Wittig reaction performed was found to yield the E- isomer of p-methoxycinnamic acid. Better yields may be obtained if recrystallised ester is hydrolysed instead of crude ester.
Acknowledgements
Due to time restrictions, I was unable to attain an IR spectrum for my own recrytallised hydrolysis product, and so ****** gave me permission to use a copy of the spectrum she obtained from her product.
[1] http://ochem.net/Wittig_reaction (accessed 30/10/2010)
[2] Clayden, Greeves, Warren, Worthers, Organic Chemistry, Oxford University Press, Oxford, 2001, pages 814-7
[3] Clayden, Greeves, Warren, Worthers, Organic Chemistry, Oxford University Press, Oxford, 2001, pages 814-7
[4] http://www.spectraonline.com/code/CmpdList.asp?Row=9 (accessed 01/11/2010)
[5]http://www.hmdb.ca/metabolites/HMDB02040 (accessed 01/11/2010)
[6] Pavia, Lampman, Kriz, Vyvyan, Introduction to Spectroscopy, Brooks/Cole Publishing, 4th (International Student) edition, 2008, pages 241-2
[7] Clayden, Greeves, Warren, Worthers, Organic Chemistry, Oxford University Press, Oxford, 2001, pages 814-7
