Investigating Substitution Reactions of Various Alcohol-Containing Compounds
By: Kristen Powell, Morgan Sanders, Robert Walker
This is a Laboratory Journal of Organic Chemistry Note for synthesis reaction data obtained in the Organic Chemistry Laboratory—All information was experimentally conducted. This article should be used as a reference when conducting your own synthesis reactions.
Three reactions containing alcohol groups, on primary and secondary carbons reacting with halide groups have been the topic of investigation through experimental testing. The substitution pathway varied between reactions due to the different structures. Variables such as the position of carbon of interest, the most substituted or least substituted carbon, dictated the reaction mechanism that would occur within the reaction schemes produced (SN1 or SN2). The following data and findings demonstrate the proper order and trends involved in the synthesis reactions tested. The results are from data that was compiled over the course of several trials of each reaction.
The reactive nature of nucleophiles has been studied for many years. Nucleophilic substitution reactions date back to the 1930’s when such mechanisms were first discovered. Even with today’s technology and the information know about much more complicated reaction schemes, basic organic synthesis remains as the foundation for almost every reaction. SN1 and SN2 are these key building block reactions. The S clarifies that it is a substitution reaction, the N stands for the nucleophilic attack, and the number on the end represents the order of the reaction. For example; SN2 reaction is a one step process. The nucleophile attacks the sp3 orbital of the carbon that the leaving group is attached to, and the leaving group leaves all at the same time. This creates a transition state when drawing out mechanisms to display all that is happening at one moment in time. A SN1 reaction occurs in two steps. The leaving group leaves, creating a carbocation intermediate, and then the nucleophile joins. Since the carbocation puts the molecule into a single plane, the nucleophile can join from either side, creating more than one possible product. This mixture of enantiomers is known to be a racemic mixture. Additional products could have also been crated based off of what carbon the leaving group leaves off of. A carbocation wants to be on the most substituted carbon. So if it is on a carbon attached to another single carbon and a neighboring carbon is attached to two carbons, then the carbocation will go through resonance forms until it settles on the most substituted carbon. This will create a more stable molecule and allow for the activation energy of the reaction to easily be met.
Basic organic synthesis is the foundation for almost every complicated mechanistic system of reaction. In order to fully understand challenging concepts, the basic synthesis mechanism must be first investigated and understood. These synthesizes include SN1, SN2. SN1 and SN2 reactions abound the organic world, and require very specific conditions in order to react. SN1 reactions occur in a stepwise fashion, creating intermediates through rearrangement, by which being limited by the step of rearrangement. SN2 reactions occur in a concerted mechanism, which allows for straight forward product creation. Both SN1 and SN2 has the nucleophile attacking at a SP3 hybridized planar system, but only SN1 will cause a non-planar intermediate. Unlike inorganic reactions, many organic reactions require a setup that allows for the successful activation energy to be met, while creating a local that allows for the proper exchange or replacement of materials that creates a usable product.
The three substitution reactions that were conducted above went through one of these substitution reactions and could have possibly gone through both. To further confirm which mechanism was utilized, a Gas Chromatograph, Infrared Spectroscopy, and 1H NMR were conducted. These instruments identify functional groups, percent composition, and the location and coupling of hydrogens.
The three reactions above contain an alcohol electrophile (leaving group) and a strong nucleophile to replace it. A variety of lab instruments and processes are conducted to get the desired product. There is a wide range of experimental paths and substitutions that carbons can undergo. Additional building blocks to more complex mechanisms include reactions such an Elimination or addition, each type utilizes different reagents with the same starting material to achieve different products. The chosen reactions were selected to focus specifically on sp3 hybridization of the alcohol-containing carbon compound.
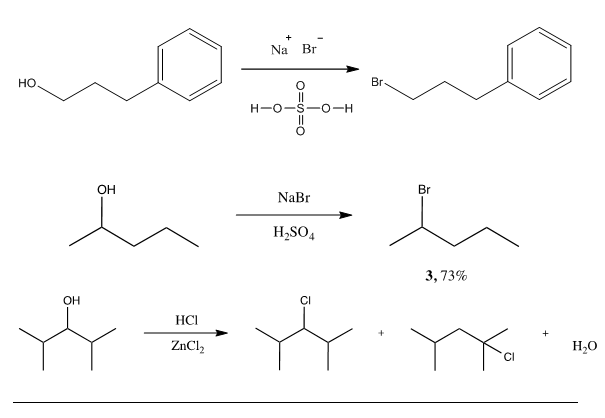
Synthesis of 1, involving a primary alcohol, yielded the above product in moderately high yield (75%) after reflux with sodium bromide and sulfuric acid (Scheme 1).
Scheme 1. Substitution of 3-phenyl-1-propanol to 1-bromo-3-phenylpropane.
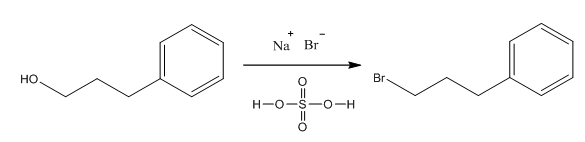
SN2 reaction occurred replacing OH group with Br halide at primary carbon location. Utilization of reaction under acidic conditions prevents E2 substitution from occurring. 1HNMR showed XXX product being formed. With substitution reactions occurring under reflux utilizing H2SO4 temperature must be observed to avoid scorching of the acid, which is indicated by a dark brown coloration occurring within the reaction.
Due to the high boiling point of these aromatic materials, care was taken to heat very gently to avoid scorching or decomposition of other reagents. No other byproducts, such as E2 elimination, often seen with more basic nucleophiles and good leaving groups, or rearranged products, as might be expected with the alternative step-wise mechanism, were observed by 1H NMR or gas chromatography. This result is consistent with a concerted substitution mechanism. Because the initial alcohol was not chiral, no stereochemical analysis was necessary. Varying the conditions (reaction temperature and equivalents of sodium bromide/sulfuric acid added) had noticeable effects on the reaction yield. However, in all reactions, using more than 1.2 eq. of NaBr resulted in no significant yield increases in this study. Omission of sulfuric acid led to very low to no yield, due to the poor nature of the leaving group. Amount of sulfuric acid added was varied slightly to ensure solubility of NaBr upon reflux. Reflux was necessary to overcome activation energy. Special note must be taken of a compound’s volatility or high boiling nature during reflux, distillation, and handling.
Scheme 2. Substitution of 2-pentanol to form 2-bromopentane
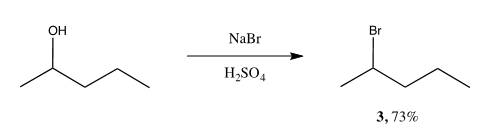
Reaction 2 combined sodium bromide and sulfuric acid with 2-pentanol to obtain the desired product. Varying amounts, conditions and procedural steps had noticeable effects on the product yielded. Refluxing and the addition of sulfuric acid were necessary in overcoming the activation energy barrier for the reaction. Without either, the compounds will not react. Due to the low boiling point of the product, reflux was conducted with caution, at minimal temperatures to avoid scorching the product. Using more than 2 eq. of NaBr resulted in no significant effect or increase of product yielded. The order of procedural steps was imperative to obtaining a high yield. Utilizing a separatory funnel before distilling could result in excess NaBr crystals interfering in the separatory funnel. The water needed to dissolve this problem decreases and almost eliminates the yield.
Analyzing the GC, IR and 1H NMR confirmed the formation of one true product. The lack of other products proves the starting material underwent SN2synthesis. SN1 would have created two different products due to the nature of the carbocation and the direction of the nucleophile joining. The nucleophile, NaBr, reacted with the poor leaving group, -OH, creating a transition state and attached itself to the carbon chain from the more open side of the molecule, opposite from the leaving group side. The first trial did not produce enough product for analysis. The data obtained is from the second trial of the experiment. The 1H NMR shows no traces of an alcohol functional group. It displays the splitting of the hydrogen attached to the carbon with the –Br appropriately, as a sextet. The IR confirms the presence of bromine and lack of –OH. The GC showed the product 100% with an insignificant presence of starting material.
Scheme 3. Substitution of 2,4,6-trimethylcyclohexanol to form 1-bromo-1,3,5-trimethylcyclohexane.
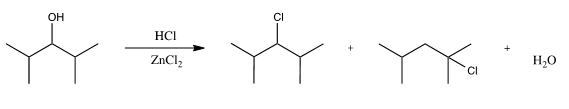
Again, varying the conditions (reaction temp. and moles of sodium bromide or sulfuric acid added) had noticeable effects on the reaction yield. However, in all reactions, using more than 1.2 eq. of NaBr resulted in no significant yield increases in this study. Omission of sulfuric acid led to very low to no yield, due to the poor nature of the leaving group. Amount of sulfuric acid added was varied slightly to ensure solubility of NaBr upon reflux. Reflux was again necessary to overcome necessary activation barriers. Extensive reflux led to the presence of eliminated products as evidenced by peaks in the 5-6 ppm range of the 1H NMR.
In conclusion, it was discovered that a selection of varied alcohol-containing compounds led predominantly to substituted halogen-containing products upon reaction with hydrogen halides. The products were formed, based on the nature of the alcohol starting material, due to the mechanism believed to occur (via SN1 or SN2). These results are only applicable to a similar selection of primary and secondary alcohols on sp3 hybridized carbons. Further study is needed to test the bounds of these substitution reactions in more extreme or unique situations/conditions.
Experimental Section
All reactions were carried out under normal atmospheric conditions. Chemicals were used directly from the manufacturer’s bottle unless otherwise mentioned. 1H NMR spectra were gathered using a Varian Gemini 200 MHz spectrometer. IR spectra were gathered using a Thermo-Nicolet 380 FT-IR. Gas chromatographs were obtained using a GOW-MAC 69-400-TCD GC (1).
3-phenyl-1-propanol (1). Added 1 ml of 6 M sulfuric acid to 3-phenyl-1-propanol (2 ml, .014 mol) to distilled 3 g Sodium Bromide in 15 ml distilled H2O (3.4 g, .033 mol). Heated under reflux at around 100 °C to prevent burning of sulfuric acid, for 60 m. Reactions occurring at excessive temperatures show dark brown residue in flask. Reaction cooled to room temperature, then utilized a separatory funnel with 30 ml dimethyl chloride solution to clean products. Organic layer formed in top of funnel, with inorganic layer on bottom. Isolated organic layer, distilled off DCM for 10 m at 60 °C to retrieve product 1 (1.32 g, .005 mol, ). 1H NMR (CDCl3, 200 MHz) δ6.9-7.2 (q,6H), 3.2-3.55 (t,3H), 2.5-2.6 (t, 3H), 2.1-2.2 (s, 2H), 1.6-1.8 (q, 3H). IR (cm-1) 3258, 3061, 3025, 2919, 2335, 2065, 1946, 1874, 1805, 1750, 1639, 1602, 1583, 1452, 1241, 1154, 1063, 918, 849, 808, 747, 573. GC (TCD) 4.5 m (less than 1%).
2-bromopentane. (2). Preparation of reaction 2 required the alcohol starting material (3.0 g, 0.034 mol) and sodium bromide (4.9 g, 0.048 mol) to be added to a round bottom flask and mixed with a catalyst, sulfuric acid (7mL of 9M). This mixture was heated under standard reflux for 30 m. During reflux, the mixture developed into layers, which are more noticeable after refluxed mixture has cooled. Both layers were an off yellow color, one more transparent than the other. Once refluxed, the resulting solution containing the product was distilled until no more would distil over; leaving a crusty liquid remaining in the round bottom flask. Careful attention was paid to the remaining reaction material so that it did not overheat and completely dry up. The distillate was then separated through an extraction process with one wash of 15 mL of water and a second wash of 5 mL of 5% NaHCO3. The organic layer was noted as the bottom layer in both washes and was then separated from the aqueous layer that contained minimal starting material and other impurities. This separation process was then followed by the addition of a drying agent, sodium sulfate. This removed any aqueous components left over from the wash. Remaining was the desired product of liquid 2 (3.8 g, 0.025 mol, 73%). The 1H NMR, IR and GC were then preformed and used to support this reaction. The results are as follows: (1H NMR spectra could not be obtained due to errors in instrument/preparation, a theoretical 1H NMR spectra from ChemDraw is utilized in place) 1H NMR (DMSO, 300 MHz) 3.80 (sext, 1H), 1.71 (q, 2H), 1.71 (d, 3H), 1.31 (sext, 2H), 0.89 (t, 3H). IR (cm-1) 2962, 2875, 1457, 1379, 1252, 1200,1147, 615. GC (TDC) 7.2 m (100%).
2,4-dimethyl-3-pentanol (3). Product 3 was prepared by adding starting material (1.78g, 0.0153mol) and Lucas reagent (5.0mL) to a 50mL round bottom flask and heating in a warm water bath (~55°C) for 20 minutes. The solution was then separated by density in a separatory funnel with the organic layer on top. The organic layer was washed with 10mL H2O twice and once with NaHCO3 (0.034g in 10mL H2O). Product 3 was collected and weighed (0.958g, 0.00824mol, 53%). To analyze the product, 1H NMR, IR, and GC were used. 1H NMR (CDCl3, 200 MHz) N/A. IR (cm-1) 2960, 2872, 1677, 1468, 1387, 1369, 1223, 1134, 990, 840, 813. GC (TCD) 4.5m (89%) 7.2m (11%)
Acknowledgement. This work was made possible by the Department of Chemistry and Chemical Biology at IUPUI.
References. Your references should be included here, not as footnotes. You should site this hypothetical paper (citation below) when writing your own article. You should also site any other references used. The number below (1) should correspond to a superscript found wherever the information was used in the article. This information should all be deleted in your final draft.
Hypothetical paper you just read:
1. Denton, R.E.; Audu, C. “Investigating Substitution Reactions of Various Alcoholic Compounds.” Fake Journal of Organic Chemistry 2010, 77, 3452-3453.
