A Study of Regiochemistry and Stereochemistry in the Hydroboration-Oxidation of (1R)-(+)--Pinene
By: Ryan DeAngelis, Andrew Mound
Introduction
The purpose of this experiment is to synthesize an alcohol using hydroboration-oxidation and determine the regiochemistry and stereochemistry of the product via 1H-NMR, 13C-NMR, and Infrared spectroscopy.1
Hydroboration-Oxidation is a two-step process used to convert an alkene to an alcohol. The first step, hydroboration, is the addition of borane (BH3) to the alkene. It proceeds as a concerted mechanism with the electron pair in the pi bond attacking the electrophilic boron, and one of the hydrogens of the borane molecule attacking the more substituted carbon. This step occurs through a cyclic, four-centered transition state.2
The second step, oxidation, involves the addition of sodium hydroxide (NaOH) and hydrogen peroxide (H2O2) to the trialkylborane formed in the first step. The H2O2 forms a negatively charged hydroperoxide ion that acts as a nucleophile and attacks the trialkylborane. A 1,2 shift of the alkyl group and oxygen results in the ejection of a hyroxide ion. This nucleophilic attack and 1,2-shift occurs two more times, resulting in a trialkylborate. The trialkylborate reacts with aqueous NaOH to form sodium borate and the final product, an alcohol.2
This experiment will focus on hydroboration-oxidation of (1R)-(+)-a-pinene. The reaction scheme for the hydroboration-oxidation of (1R)-(+)-a-pinene is shown below.
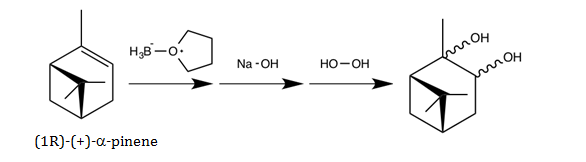
Figure 1: Hydroboration-Oxidation of (1R)-(+)-a-pinene.
The product in Figure 1 shows the OH group bonded to either carbon 2 or 3 and does not specify spatial orientation. Consequently, there are eight products that could theoretically form. The starting material can undergo hydroboration in a Markovnikov or anti-Markovnikov fashion, and this can occur from either the top or bottom face of the molecule. Markovnikov addition means the hydrogen adds to the least substituted carbon, and anti-Markovnikov means the hydrogen adds to the most substituted carbon.3 Further, the oxidation step can occur in a syn-addition or anti-addition fashion. Syn-addition is when two molecules add to the same face of the molecule, and anti-addition is when two molecules add to opposite faces of the molecule.3 Thus, by considering all possible regioselectivity and stereoselectivity combinations of the reaction, there are eight possible isomers.
However, understanding the hydroboration-oxidation mechanism allows for an educated guess as to which isomer is most likely to form. The hydroboration step occurs first with the boron and hydrogen adding to the same face via syn-addition. The hydrogen is bonded to the boron, and therefore, is not able to add anti-addition. The boron adds in an anti-Markovnikov fashion due to steric hindrance; the boron is much larger than the hydrogen atom, and thus, needs more “space.” Therefore, the boron bonds to the less substituted carbon. The addition of the borane most likely occurs from the bottom face of the molecule, due to the steric hindrance created by the bridgehead carbon and the two attached methyl groups. In the oxidation step, there is retention of stereochemistry due to the trialkyklborate simply reacting with aqueous NaOH. Although an understanding of the mechanism and previous organic knowledge allows for a well-educated guess, whether or not this previous knowledge of hydroboration-oxidation applies to sterically-hindered, bicyclic ring systems will be determined.
In terms of “real-world” application, hydroboration-oxidation is particularly useful due to its extremely high stereoselectivity and regioselectivity. Additionally, due to the cyclic, four-centered transition state formed during the hydroboration step, rearrangement is not possible, being that no carbocation is formed.4
Experimental Procedure
The experimental procedure was very similar to the one outlined in class, with some minor modifications.1
309 mg of (1R)-(+)-a-pinene was weighed directly into a 10 mL round-bottom flask. The flask was then equipped with a Claisen head containing a CaCl2 drying tube, rubber septum, and stir bar. BH3 THF was added directly to the round-bottom flask using a dry syringe through the septum. The mixture was heated while stirring at 50 degrees Celsius for ten minutes. The reaction was cooled using an ice water bath, followed by the addition of two drops of water. Once the frothing stopped, four more drops of water were added. A reflux condenser was attached to the round-bottom flask, and 750 mL of 3.0 M NaOH was added via an automatic delivery pipette. Shortly thereafter, 750 mL of 30% H2O2 was added via an automatic delivery pipette. The mixture was heated to a gentle reflux for 20 minutes. After heating for 20 minutes, the reaction was cooled using an ice bath, and the aqueous layer was extracted via a pipette to a 5 mL vial. The vial and flask were labeled, and both the aqueous and organic layers were stored in the refrigerator until the following lab period.
In the following week, both the aqueous and organic layers were acquired from the fridge. 3 mL of petroleum ether was added to the aqueous layer, and two layers formed. A small organic layer was extracted from the aqueous layer vial. 4 mL of 0.20 M HCl was added to the organic layer, and the organic layer was extracted. The organic layer was washed with two times with 2 mL of deionized water. The organic layer was dried on Na2SO4, and added to a 125 mL filter flask.
A 15 mL glass centrifuge tube was cleaned and dried before being placed into a rubber stopper. The sublimation apparatus was assembled placing the tube-stopper piece into the 125 mL filter flask until the tube was about 0.5 cm from the bottom. The filter flask was connected to the vacuum line via a rubber hose and a vacuum was applied to the system. Once a vacuum was established, dry ice was carefully added to the centrifuge tube via forceps, and a careful effort was made to not allow any water to enter the system. The bottom of the filter flask was heated using a micro burner, and heat was continuously applied as the product sublimated. When the sublimation was complete, the heat source was removed, but the aspirator was not. The flask cooled under reduced pressure while crystals formed on the bottom of the centrifuge tube. The system was vented once cooling completed. The centrifuge tube was removed and the product was scraped off the sides of the centrifuge tube and flask. CDCl3 was added to the flask in order to dissolve the product that was not scraped from the sides of the flask. A portion of the product was immediately added to an NMR tube, and the remaining product was placed in a vial and labeled. 1H-NMR, 13C-NMR, and IR spectroscopy of the product were acquired. The lab area was cleaned and all equipment was put away.
See attached page for calculations of mmol used, theoretical yield, and a flow chart for the “Work-up and Isolation of the Crude Product.” Although product was isolated, a mass was not acquired due to the product being dissolved in CDCl3, and thus, there was no “actual yield” calculation.
Results/Data/Spectra
Table 1: 1H-NMR (CDCl3) Key Peak Table for (1R)-(+)-a-Pinene
|
Chemical Shift (ppm) |
Type of Signal |
J Values (Hz) |
Integration |
Type of Proton |
|
0.83 |
S |
—- |
3 |
CH3 |
|
1.14 |
D |
11.20 |
1 |
CH |
|
1.25 |
S |
—- |
3 |
CH3 |
|
1.93 |
T of D |
1.60 |
1 |
CH |
|
2.06 |
M |
—- |
—- |
—- |
|
2.19 |
M |
—- |
—- |
—- |
|
2.33 |
D of T |
10.80 |
1 |
CH |
|
5.18 |
—- |
2.00 |
1 |
C=CH |
*Note: these are the key peaks readily identifiable in the spectrum
Table 2: 1H-NMR (CDCl3) Peak Table for Hydroboration Product
|
Chemical Shift (ppm) |
Type of Signal |
J Values (Hz) |
Integration |
Type of Proton |
|
0.90 |
S |
—- |
3 |
CH3 |
|
1.11 |
D |
7.36 |
3 |
CH3 |
|
1.20 |
S |
—- |
3 |
CH3 |
|
1.71 |
D of D of D |
13.96, 4.72, 2.60 |
1 |
CH |
|
1.78 |
T of D |
5.96, 1.96 |
1 |
CH |
|
1.93 |
M |
—- |
2 |
CH2 |
|
2.35 |
M |
—- |
1 |
CH |
|
2.50 |
M |
—- |
1 |
CH |
|
4.05 |
D of T |
9.48, 5.08, 4.96 |
1 |
CH |
Table 3: 13C-NMR (CDCl3) DEPT Experiment Peak Table for Hydroboration Product
|
Chemical Shift (ppm) |
Type of Carbon |
|
|
20.8 |
CH3 |
|
|
23.7 |
CH3 |
|
|
27.7 |
CH3 |
|
|
34.4 |
CH2 |
|
|
38.2 |
C |
|
|
39.0 |
CH2 |
|
|
41.8 |
CH |
|
|
47.7 |
CH |
|
|
47.8 |
CH |
|
|
71.6 |
CH |
|
Table 4: Infrared Spectroscopy Peak Table for 4-tert-butylcyclohexanone
|
Absorption (cm-1) |
Functional Group |
|
3025.51 |
Alkenyl C-H Stretch |
|
2986.06, 2879.98, 2915.92 |
Alkane C-H Stretch |
Table 5: Infrared Spectroscopy Peak Table for Hydride Reduction Product
|
Absorption (cm-1) |
Functional Group |
|
3271.60 |
Alcohol O-H Stretch |
|
2988.86, 2901.42 |
Alkane C-H Stretch |
Due to the final product being dissolved in CDCl3, no melting point measurement was acquired.
Discussion
In analyzing the 1H-NMR of (1R)-(+)-α-pinene, certain key peaks are readily identified (see attached spectrum). The two singlets present at 0.83 ppm and 1.25 ppm are identified as H9 and H8, respectively. Both have an integration of three, but the H8 protons are deshielded due to their proximity to the double bond. The H9 protons are further from the double bond, and therefore occur farther upfield. The second key peak is the H3 alkenyl proton at 5.18 ppm. This proton couples with the two neighboring diastereotopic protons to form a doublet of doublets.5 The peaks observed at 1.14, 1.93, 2.06, 2.19, and 2.33 ppm cannot be accurately identified in the given spectrum.
In analyzing the 1H-NMR of the hydroboration product, the singlet at 0.90 ppm is the H9 protons, and the singlet at 1.20 ppm is the H8 protons. Similar to the starting material, the H8 protons are farther downfield due to the deshielding caused by the electron-rich oxygen atom on carbon 3.5 The doublet at 1.11 ppm is identified as the H10 protons. The H10 protons couple to the proton on carbon 2, and the peak is shifted slightly downfield due to the neighboring oxygen atom.5 The peak downfield at 4.05 ppm is the H3 proton. This peak is a doublet of triplets, and is coupled with the H2, H4, and H4’ protons. It is shifted significantly downfield due to oxygen atom being bonded to the same carbon. The peaks observed between 1.71 ppm and 2.50 ppm cannot be accurately identified in the given spectrum.
Infrared (IR) spectroscopy is particularly useful in determining the functional groups present in a molecule, and thus, is extremely helpful in determining whether the hydroboration product was created. The IR spectrum of the starting material has a characteristic peak at 3025.51 cm-1, representing an alkene C=C-H stretch (see Table 4).4 In the IR spectrum of the hydroboration product, there is no peak in this region, which in turn represents the loss of this double bond. However, the appearance of a broad peak at 3271.60 cm-1 is characteristic of an alcohol O-H stretch (see Table 5).4 Thus, the appearance of this alcohol O-H stretch indicates that the desired hydroboration product was created.
In theory, there are eight possible isomers that could form in the hydroboration-oxidation of (1R)-(+)-α-pinene. However, four isomers can immediately be eliminated. There are four possible Markovnikov products and four possible anti-Markovnikov products. When looking at the Markovnikov products, it is apparent that two of the structures are identical, and thus, two of the structures can be eliminated. For the anti-Markovnikov products, the two products where the hydroboration occurs from the top face can be eliminated. This addition does not occur from the top of the molecule due to the presence of the bridgehead carbon. The bridgehead carbon has two methyl groups attached, and thus, creates steric hindrance above the molecule. As a result, the hydroboration must occur from the bottom face of the molecule. Accordingly, the four remaining isomers are the Markovnikov and anti-Markovnikov products where the hydroboration occurs from the bottom face.
In order to determine whether the reaction involves anti-Markovnikov or Markovnikov addition, the 1H-NMR must be analyzed. The main difference between these two products is the groups attached to carbons 2 and 3. In the Markovnikov product, the protons on the methyl group attached to carbon 2 appear as a downfield-shifted singlet, and the carbon 3 protons split into a triplet of doublets.5 The carbon 3 protons couple with the other proton on carbon 3 and the two neighboring diastereotopic protons on carbon 4.5 In the anti-Markovnikov product, the protons on the methyl group attached to carbon 2 split into a doublet due to the proton present on carbon 2. The carbon 3 protons couple with the carbon 2 proton, as well as the neighboring diastereotopic protons on carbon 4, to form a doublet of triplets.5 When examining the 1H-NMR spectrum, it is clear that the anti-Markovnikov product formed, due to the presence of the aforementioned characteristic peaks (see labeled class hydroboration product spectrum).
The final hydroboration product contains 10 total carbons. The anti-Markovnikov product contains four CH carbons, three CH3 carbons, two CH2 carbons, and one quaternary carbon. The Markovnikov product contains two CH carbons, three CH2 carbons, three CH3 carbons, and two quaternary carbons. The 13C-NMR has 10 total peaks, one for each of the carbons present in the product. The DEPT experiment at 1350 has nine peaks, which shows that there is only one quaternary carbon present in the product. Additionally, the DEPT experiment at 1350 shows the presence of two CH2 carbons. The DEPT experiment at 900 shows the presence of four CH carbons, and by cross-referencing this DEPT experiment with the DEPT experiment at 1350, it can be determined that there are three CH3 carbons. Based on the number and type of specific carbons present as determined by the DEPT experiments, it is clear that the anti-Markovnikov product formed.
Based on the splitting trees constructed for proton H3 for Alcohols 2 and 3 (see attached), it is clear that Alcohol 2 formed. The splitting pattern for Alcohol 2 is a doublet of triplets, and the splitting pattern for Alcohol 3 is a triplet of doublets.6 In examining the 1H-NMR of the product, the H3 peak is a doublet of triplets. The splitting tree does not look identical to the actual splitting in the spectrum because the two inside peaks overlap one another and cause a larger, broader peak to form. According to the coupling constants table, proton H3 couples with H2, H4’, and H4 with coupling constant values of 5.0 Hz, 8.5 Hz, and 5.0 Hz, respectively.6 In the class spectrum, the coupling constants for H3 coupling with H2, H4’, and H4 were determined to be 5.08 Hz, 9.48 Hz, and 4.96 Hz, respectively. In the personal spectrum, these values were determined to be 4.92 Hz, 9.68 Hz, and 5.48 Hz, respectively. The personal 1H-NMR spectrum was not as pure as the class 1H-NMR spectrum, and thus, only the coupling constant for the doublet created by the H9 protons was calculated. The H10—H2 coupling constant calculated in the personal spectrum was 7.80 Hz, and for the class spectrum, this value was 7.36 Hz. The coupling constants for the class spectrum and personal spectrum are similar to one another, and these values are also similar to the literature values.6 This indicates that the same compound was formed.
Conclusion
The purpose of this experiment was to use various spectroscopic techniques to determine the regiochemistry and stereochemistry of an alcohol synthesized via hydroboration-oxidation.1 After extensive analysis of 1H-NMR, 13C-NMR, and Infrared spectroscopy, it is clear that Alcohol 2 formed. The reaction occurred via anti-Markovnikov, syn-addition from the bottom face of the starting material. Thus, the hypothesis was correct, and previous organic knowledge of hydroboration-oxidation applied to a sterically-hindered, bicyclic ring system. Future work could include a proton-decoupling or COSY experiment to further identify peaks in the 1H-NMR spectrum.
References
- “Hydroboration-Oxidation of (1R)-(+)-α-Pinene,” Dr. Lynn Bradley, The College of New Jersey.
- Brown, William, Christopher Foote, Brent Iverson, and Eric Anslyn. Organic Chemistry. 5th ed. Brooks/Cole, 2008. Print.
- Loudon, Marc. Organic Chemistry: Addition Reactions of Alkenes. 4th ed. New York: Oxford University Press, 2002. Web.
- Vollhardt, Peter, and Neil Shore. Organic Chemistry: Structure and Function. 5th ed. New York: W.H. Freeman and Company, 2007. Web.
- “Lecture Notes on 1H-NMR,” Dr. Lynn Bradley, The College of New Jersey.
- “Stereoisomers of (1R)-(+)-α-pinene and Coupling Constants for H3 in Alcohols 2 and 3,” Dr. Lynn Bradley, The College of New Jersey.
