Synthesis of Dihydrojasmone
By: Robert Burns
Abstract:
The purpose of this experiment was to successfully synthesize dihydrojasmone through a series of synthetic steps starting from levulinic acid. Then, using 1,1’-carbonyldiimidazole (CDI), the acid was turned into a Weinreb amide. After a simple conversion of the carbonyl group to a ketal, the amide was reacted with hexyl magnesium bromide to give the 1,4 diketone. The diketone was then refluxed under basic conditions to afford dihydrojasmone as a light brown liquid (0.06g, 28.0%). All products were characterized by NMR and IR spectroscopy.
Introduction:
Dihydrojasmone is a compound which contains a very strong fragrance and has many applications in the fragrance industry as well as adding aroma to different substances. It is a key aroma compound related to the hop aroma characteristics in beer selected by PCA and is fruity, sweet, floral, and woody with a powdery nuance8. Dihyrdrojasmone will be synthesized in the lab starting with levulinic acid. The first step begins with reacting the levulinic acid with CDI and N,O-dimethylhydroxylamine hydrochloride to afford the Weinreb amide through a carbamate intermediate4. The next step of the synthesis involves reacting the levulinic amide with p-toluenesulfonic acid, trimethyl orthoformate, and methanol, simply converting the carbonyl group to a ketal. The next step is involves reacting the carefully dried amide with hexyl magnesium bromide to give the diketone through a chelated intermediate. “We believe that this conversion procedes through a very stable metal-.chelated intermediate, which probably accounts for the observed lack of over-addition products”6. The final step of the synthesis is a simple aldol reaction under basic conditions, turning the diketone into dihydrojasmone.
This synthetic route was chosen because of the simplistic yet elegant features. The starting materials and reagents were also relatively cheap making this a practical synthetic route. Another synthetic route proposed by Shindo7 involves using γ-keto esters or keto diesters as suitable substrates for this tandem reaction to provide synthetically useful 2,3- disubstituted cyclopentenones. The synthesis of dihydrojasmone involves reacting the pentyl-substituted ynolate with ethyl 4-oxopentanoate to give the product with an 80% yield. The only aspect in the Shindo synthesis which may make it impractical is the use of t-BuLi, which is extremely pyrophoric and dangerous.
Discussion:
The majority of the reactions gave average yields with fairly good purity as seen in the NMR spectra of the products. The expected peaks are seen in the spectra although some contain solvent and impurities as well. Because each of the starting reagents significantly differs from the product in polarity, column chromatography using silica gel was employed for most purification steps. This purification step was crucial before starting the Grignard due to the fact that hexyl magnesium bromide does react with water was well as carbonyl containing compounds.
The overall reaction scheme is as follows
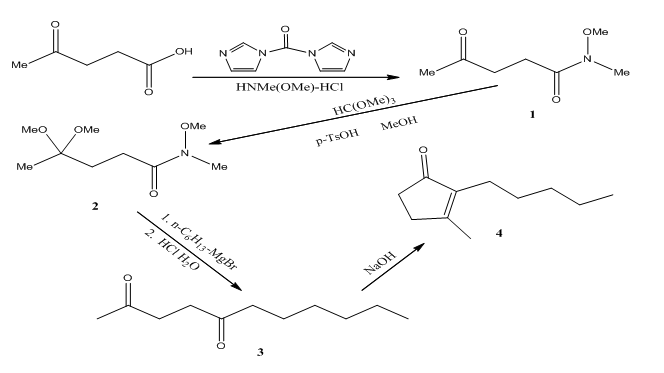
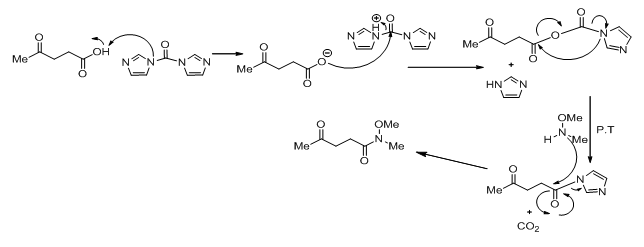
The conversion of levulinic acid to 1 is done using CDI which converts the acid to a carbamate and then releases imidazole and carbon dioxide. This intermediate then reacts with N ,O-dimethylhydroxylamine hydrochloride to give the Weinreb amide.
The conversion of the amide 2 to the diketone 3 is done using hexyl magnesium bromide through a magnesium chelated intermediate. After acidic work up, the ketal is removed and the diketone is obtained.
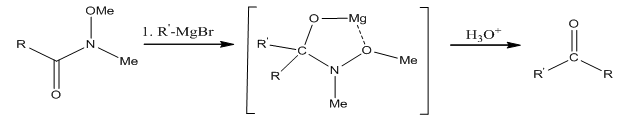
The conversion of the amide 2 to the diketone 3 is done using hexyl magnesium bromide through a magnesium chelated intermediate. After acidic work up, the ketal is removed and the diketone is obtained.
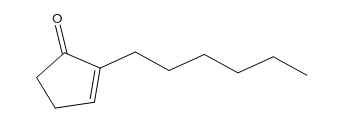
However, because of thermodynamics, dihydrojasmone is favored due to the tetra substituted alkene opposed to this tri substituted alkene.
Conclusion and Summary:
The experiment was completed with all of the products being obtained with fair yields. The purity for some of the products however was lacking. Although the NMR indicated that the products were obtained, many small side reaction impurities were present. These could have been avoided by trying to weigh reagents more carefully or not over heating a reaction. The column chromatography using silica gel seemed to reduce impurities by a significant amount and should be used for subsequent purification.
Experimental:
Synthesis of N-methoxy-N-methyl-4-oxopentamide
Levulinic acid (5.00 g, 43 mmol) and methylene chloride (90mL) are added to a 200 mL round bottom flask containing a stir bar. Then 1,1’-carbonyldiimidazole (6.98 g , 43 mmol) was slowly added to the round bottom flask. After the solution was stirred at room temperature for ten minutes under nitrogen, N,O-dimethylhydroxylamine hydrochloride (4.23 g, 43 mmol) was added. The reaction was stirred overnight under nitrogen at room temperature and then diluted with methylene chloride (50 mL). The solution was then washed with 0.5 N HCl ( 2 x 100 mL), saturated sodium carbonate (2 x 100 mL), and saturated sodium chloride (100 mL). The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure to give N-methoxy-N-methyl-4-oxopentamide 1 as a clear liquid (3.87g, 56.5%).
Synthesis of N,4,4-trimethoxy-N-methylpentanamide
The levulinic amide (3.9 g, 24.5 mmol), p-toluenesulfonic acid (0.12 g, 0.7 mmol), trimethyl orthoformate (15 mL) and methanol (25 mL) were combined in a 100 mL round bottom flask containing a stir bar. The reaction was then refluxed under nitrogen for 3 h and left at room temperature overnight. The reaction was then diluted with diethyl ether (200 mL) and washed with 5% sodium hydroxide solution (100 mL). The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure to give 4,4-dihydroxy-N-methoxy-N-methylpentanamide 2 as a yellow liquid (0.57g, 11.3%). The product contained many impurities and was chromatographed using silica gel.
Synthesis of Undecane-2,5-dione
A 100 mL round bottom flask containing a stir bar was charged with the dihydroxy N-methoxy-N-methylpentanamide (0.57g, 2.77 mmol) and 30 mL of anhydrous THF under nitrogen. The reaction was placed into an ice bath and hexyl magnesium bromide (4.20 mL, 4.16 mmol) was added slowly while stirring. The reaction was allowed to stir for 1 h and was monitored by TLC ethyl acetate/hexanes (1:4). The reaction was then quenched with 20 mL of 0.5 N HCl and allowed to stir overnight at room temperature. The reaction was then diluted with 20 mL of methylene chloride and washed with saturated sodium bicarbonate (2 x 20 mL) and saturated sodium chloride (15 mL). The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure to give the undecane-2,5-dione 3 as a viscous yellow oil (0.22g, 43.3%). The diketone was then subject to chromatography using silica gel.
Synthesis of 3-methyl-2-pentylcyclopent-2-enone
A 100 mL round bottom was charged with diketone (0.22g, 1.20 mmol), 2% NaOH solution (20 mL), and ethanol (4mL). The mixture was then refluxed for 6 h and diluted with water (20 mL). The mixture was washed with diethyl ether (2 x 20 mL), the organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure to give the 3-methyl-2-pentylcyclopent-2-enone 4 a light brown liquid (0.06g, 28.0%).
N-methoxy-N-methyl-4-oxopentamide spectra data
1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 2.73 (t, J = 5.2 Hz, 2H), 2.71 (s, 3H), 2.50 (t, J = 5.2 Hz, 2H), 2.13 (s, 3H).
13C NMR (100 MHz, CDCl3) δ 207.2, 172.8, 60.9, 53.4, 37.2, 31.9, 29.7, 25.7.
IR (neat, cm-1) 2911, 2843, 1715, 1657, 1418, 1387, 1165, 997.
Rf 0.56 (hexanes/ethyl acetate, 4:1)
N,4,4-trimethoxy-N-methylpentanamide spectra data
1H NMR (400 MHz, CDCl3) δ 3.63 (s, 3H), 3.11 (s, 6H), 2.71 (s, 3H), 2.41 (t, J = 8 Hz, 3H), 1.89 (t, J = 8 Hz, 3H), 1.21 (s, 3H).
13C NMR (100 MHz, CDCl3) δ 207.4, 101.0, 61.0, 60.2, 50.4, 47.8, 37.3, 30.9, 29.9, 25.8, 20.8, 20.7, 14.0.
IR (neat, cm-1) 2946, 2839, 1670, 1446, 1355, 1193, 1094, 1065, 1029, 983.
Rf 0.67 (hexanes/ethyl acetate, 4:1)
Undecane-2,5-dione spectra data
1H NMR (400 MHz, CDCl3) δ 2.73 (t, J = 7.2 Hz, 3H), 2.40 (t, J = 5.6Hz, 2H), 2.13 (s, 3H), 1.53 (pent, J = 5.6 Hz, 2H), 1.31 (sex, J = 9.1 Hz, 2H), 1.29 (pent, J = 5.6 Hz, 2H), 0.88 (s, 3H).
13C NMR (100 MHz, CDCl3) δ 209.4, 207.1, 176.6, 170.9, 62.7, 60.2, 42.6, 40.8, 36.7, 32.8, 32.6, 31.7, 29.7, 28.7, 25.4, 25.3, 23.6.
IR (neat, cm-1) 2956, 2928, 2858, 1740, 1713, 1459, 1402, 1371, 1240, 1162, 1083, 1046.
Rf 0.77 (hexanes/ethyl acetate, 4:1)
Dihydrojasmone spectra data
1H NMR (400 MHz, CDCl3) δ 3.65 (t, J = 7.2 Hz, 2H), 2.41 (t, J = 5.8 Hz, 2H), 2.08 (t, J = 5.6 Hz, 2H), 1.97 (s, 3H), 1.29 (pent, J = 5.6 Hz, 4H), 1.27 (sex, J = 9.1 Hz, 2H), 0.81 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3) δ 209.5, 170.9, 169.8, 140.6, 66.6, 60.2, 31.6, 31.3, 27.9, 22.8, 22.3, 20.9, 15.0, 14.0, 13.9.
IR (neat, cm-1) 2956, 2926, 2857, 1696, 1645, 1458, 1443, 1358, 1073, 730.
Rf 0.69 (hexanes/ethyl acetate, 4:1)
- Bellina, F.; Ciucci, D.; Rossi, R.; Vergamini, O. Tetrahedron. 1999, 55, 2103.
- Callejón, R.; Morales, M. L.; Troncoso, A.; Ferreira, A. J. Agric. Food Chem. 2008, 56, 6631.
- J.-L. Moreau, R. Couffignal, R Arous-Chtara Tetrahedron 1981, 22, 3815.
- P.D. Theisen, C.H. Heathcock J. Org. Chem. 2001, 53, 2374.
- R. A. Ellison, Synthesis 1973, 397.
- S. Nahm, S. M. Weinreb Tetrahedron Lett. 1981, 22, 3815.
- Shindo, M.; Sato, Y.; Shishido, K. J. Org. Chem. 2001, 66, 7818.
- Takako, I.; Fumihiko, T.; Mariko, I.; Kaneo, O. J. Agric. Food Chem. 2013, 61,4578.
