Synthesis and Properties of a Cobalt Cage Complex
Written by Dan Jenkinson
Introduction
A ‘cage’ complex is a type of molecule, where a metal ion is encapsulated in a ligand and is unable to get out. The inability for the metal ion to get out of the cage complex also means that it is unable to get into it. As such, cage complexes often have to be assembled around the metal centre in situ.
There are many examples in cage complexes, for example metals can become encapsulated within a fullerene. These types of molecules are not easy to make though, and have to be constructed using laser-induced vapour phase reactions between carbon and the metal to give a metal cation surrounded by a fulleride (a reduced fullerene).[1]
In this experiment, cage complexes that can be derived from simple bidentate ligands such as ethylenediamine (1,2-diaminoethane) are explored, specifically the ligand dinosar (1,8-dinitro-3,6,10,13,16,19-hexaazabicyclo(6.6.6)icosane).
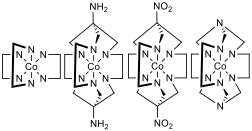
Figure 1 – Far left: [Co(en)3]3+ where en = ethylenediamine, mid left: [Co(sar)]3+ where sar = sarcophagine, mid right: [Co(dinosar)]3+, far right: [Co(sep)]3+ where sep = sepulchrate, note that the protons bonded to the nitrogens that are bonded to the cobalt centre are not displayed for clarity
These types of ligands can render metal centres that are usually very reactive (such as Co (II)) inert. Co (II) is a d7 metal centre, which makes it labile because it has less crystal field stabilisation energy.[2] When it is trapped in a cage complex, it becomes inert to substitution, simply because it cannot escape from the rest of the compound. This property if the cage complex is explored further in this experiment.
The synthesis of [Co(diNOsar)]3+ involves two reactions, the first of which is the transformation of each of the amine nitrogens bonded to the Cobalt into imines by reacting with an aldehyde or a ketone. Unless the desire of the manufacturer is to have a substituent on the newly formed C=N double bond, formaldehyde should be used as the carbonyl. The second step (encapsulation of the metal centre) is only possible due to a phenomenon called tautomerisation. Tautomerisation involves the migration of a proton, and the switching of a pair of adjacent single and double bonds. Tautomerisation is possible in highly energetic nitro compounds to their tautomers (aci-nitro groups).[3] In this case, nitromethane can tautomerize to its aci-nitro equivalent and react with the polar imine groups in the diNOsar precursor.
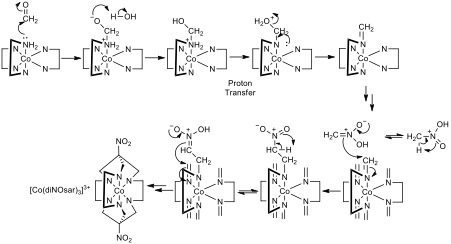 Figure 2- The mechanism for the transformation of [Co(en)3]3+ to [Co(diNOsar)3]3+, note that two arrows pointing in the same direction indicates that the same process is repeated at the other reactive centres, also note that not all amine protons are shown to reduce clutter so that mechanistic steps can be seen clearly
Figure 2- The mechanism for the transformation of [Co(en)3]3+ to [Co(diNOsar)3]3+, note that two arrows pointing in the same direction indicates that the same process is repeated at the other reactive centres, also note that not all amine protons are shown to reduce clutter so that mechanistic steps can be seen clearly
The aim of this experiment was to assemble a cobalt cage complex in situ by using smaller, bidentate ligands, and to compare its spectroscopic data to that of stock solutions of the starting material and the product.
Experimental
A water bath was prepared and maintained at a temperature between 35-40 °C. A sample of [Co(en)3]Br3 (2.56 g, 10.7 mmol) was dissolved in water (90 ml) along with anhydrous sodium carbonate (1.57 g, 14.8 mmol). To the mixture was then added formaldehyde (37% v/m, 40 ml, 0.50 mol) and nitromethane (5 ml, 93.1 mmol). The mixture was stirred for one minute before being placed in the water bath. Over the 1.5 hour incubation period the reaction was stirred three times at regular intervals, and the colour of the solution changed from orange to a dark shade of red/brown. The solution was allowed to cool to room temperature, and ethanol was added to aid precipitation of the product. The solid produced was collected with vacuum filtration, washed with ethanol then diethyl ether, and then air dried to give [Co(diNOsar)]Br3 (1.21 g, 1.97 mmol, 16.8 %). The product was of sufficiently good quality to proceed with further analysis. Solutions of [Co(en)3]Br3, bought [Co(diNOsar)]Br3 and synthesised [Co(diNOsar)]Br3 in 0.1 M sodium chloride, each with concentrations of 5 mM. UV-vis spectra and cyclic voltammograms were recorded for each one.
The conditions used for the cyclic voltammogram were as follows:
- Start potential 0 mV
- End potential -800 mV
- Upper limit 0 mV
- Lower limit -800 mV
- Scan rate 100 mVs-1
Results and Discussion
UV-Vis spectra and voltammograms can be found in the following locations
- Stock [Co(en)3]Br3 solution, UV-Vis– appendix 1
- Stock [Co(diNOsar)]Br3 solution, UV-Vis– appendix 2
- Synthesised [Co(diNOsar)]Br3 solution, UV-Vis – appendix 4
- [Co(diNOsar)]Br3, voltammogram – appendix 5
- [Co(sep)]Br3, voltammogram – appendix 6
- [Co(en)3]Br3, voltammogram – appendix 6
Table 1 – UV-Vis data
|
Appendix |
Species |
Absorption maximum |
Electronic transition |
|
|
Wavelength / nm |
Absorption |
|||
|
1 |
[Co(en3)]Br3 |
466.00 |
0.375 |
T2 à E |
|
338.50 |
0.345 |
T2 àA2 |
||
|
2 |
Stock [Co(diNOsar)]Br3 |
475.00 |
0.739 |
T2 à E |
|
343.50 |
0.635 |
T2 à A2 |
||
|
4 |
Synthesised [Co(diNOsar)]Br3 |
494.00 |
0.284 |
T2 à E |
|
331.00 |
0.606 |
T2 à A2 |
||
As well as the starting material ([Co(en)3]Br3]), two mole equivalents of nitromethane and six mole equivalents of formaldehyde were required for this reaction; however, due to the quantities used, the start material was still the limiting reagent of the reaction, and the theoretical yield of the reaction, assuming a 1:1 molar ratio of start material to product, was 7.20 g. Given this value, the yield of the reaction was 16.8 %. This is low compared to the yield stated in the literature (55 %[4]). This may be due to ineffective heating from the water bath (which may have over- or under-heated the reaction), loss during transfer of the reaction solution, or not adding enough ethanol to ensure the complete precipitation of the product from the reaction mixture.
In the preparation of the diNOsar ligand, the central metal ion plays some crucial roles. Firstly, it acts as a collecting point for reactants. Having the ethylenediamine ligands all coordinated to one metal centre makes the construction of the cage ligand much easier than if they were free in solution. The metal provides the correct stereochemical requirements, i.e. two trigonal faces of an octahedron, to build around in the construction of diNOsar.
Usually imines in aqueous solution are susceptible to hydrolysis. The first step of the aqueous hydrolysis of an imine involves the protonation of the imine in order to increase the polarity of the C=N double bond, thus enhancing the rate of nucleophilic attack from hydroxide ions. This, however, can only be done if the lone pair of the nitrogen is available for donation in order to protonate the molecule. The metal ion in this synthesis stabilises the intermediate against imine hydrolysis because nitrogen’s lone pair is datively bonded to cobalt.
A final role of the metal ion in this mechanism is that it activates the intermediate imines towards nucleophilic attack. Metals are generally electron donating, and so electron density is pushed onto the electronegative nitrogen atoms. This increases the polarity of the C=N double bond, making it slightly susceptible to nucleophilic attack from good nucleophiles such as ammonia or nitromethane. This effect must be shared over all six nitrogen atoms covalently bonded to the cobalt centre, thus the effect is lessened. As such, the imines are not vulnerable to hydrolysis, since the hydroxide ion is a poor nucleophile compared to the aforementioned nitrogen based nucleophiles.[5]
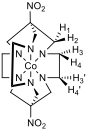
 Figure 3 – Left: proton numbering scheme for [Co(diNOsar)]3+, note that the protons bonded to the amine nitrogens are not shown since they rapidly exchange with D2O and give broad peaks with high chemical shifts which are difficult to analyse, right: 1H NMR spectrum of [Co(diNOsar)]3+
Figure 3 – Left: proton numbering scheme for [Co(diNOsar)]3+, note that the protons bonded to the amine nitrogens are not shown since they rapidly exchange with D2O and give broad peaks with high chemical shifts which are difficult to analyse, right: 1H NMR spectrum of [Co(diNOsar)]3+
Figure 3 shows the proton NMR of the synthesised cage complex. It can be noted that there are four peaks, but at first glance of the structure, there appear to only be two sets of equivalent protons. The fact that four peaks in the NMR spectrum is due to the fact that these protons are all diastereotopic. Diastereotopic groups are chemically inequivalent, can be distinguished by systems that cannot tell left from right, and have different chemical shifts in the NMR spectrum.[6]
A test to see if a CH2 group is diastereotopic is simple. If one of the protons is substituted for a deuterium, and the product shows a chiral carbon (fig 4), then the protons on the CH2 group are diastereotopic, and will have different chemical shifts in a proton NMR.
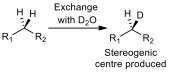
Figure 4 – A schematic of the substitution test for diastereoisomerism
In the structure of diNOsar, protons 1 and 2 can ‘see each other,’ but can see no other protons. Since they are chemically inequivalent, they cause spin-spin coupling through two bonds, and two doublets (peaks A and C) are observed in the spectrum.
Proton 3 can ‘see’ proton 4, which is two bonds away, and so the peak is split. Proton 3 can also ‘see’ protons 3’ and 4’. Proton 3’ is chemically equivalent to proton 3, so no splitting occurs, but protons 3 and 4’ are chemically inequivalent, and are three bonds apart. As such, splitting occurs in the peak produced due to proton three, but this splitting constant, 3J3-4’is different to the first splitting constant 2J3-4, and hence a more complicated peak is observed. One might expect a pair of doublet of doublets to appear in the spectrum (one each for protons 3 and 4), however, due to the fact that second-order splitting takes place, the peaks are messy. Peaks B and D are caused by protons 3 and 4.
All three solutions which had a UV-Vis spectrum recorded had cobalt in its +3 oxidation state, and hence had six d electrons. The ligating atoms in these complexes (all nitrogen) are mildly strong field, and hence the octahedral crystal splitting is low spin (since the energy difference between the T2g level and the Eg levels, Δo, is bigger than the energy penalty incurred when putting two electrons of opposing spin into the same orbital, fig 5).
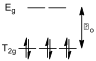
Figure 5 – The crystal field splitting diagram for a low spin d6 system
The highest occupied molecular orbital (HOMO) of this system has T2 symmetry, and there are no unpaired electrons, meaning that these electrons are in a singlet state. Photochemically driven electronic transitions can only occur from one singlet state (the HOMO) to other singlet states of higher energy. A Tanabe-Sugano diagram for the d6 system shows that there are only two other singlet states with a higher energy than the T2 level (the E and A2 levels) and that a transition from the T2 level to the E level requires less energy than a transition to the A2 level.[7] As such, two absorption maxima are expected in the UV-Vis spectra of these compounds.
Appendix three shows an overlay of the UV-Vis spectra of the stock solutions of [Co(en)3]Br3 and [Co(diNOsar)]Br3. Each one shows two absorption maxima, and it’s clear to see that the absorption maxima of the two compounds are very similar, which makes sense since the central metal ion is in the same oxidation state, and the ligating atoms are almost chemically identical. The only difference between the spectra is that the absorption values at these wavelengths are larger for the cage complex, but for each species, the absorbance was similar at each wavelength.
The UV-Vis spectrum of the synthesised cage complex also showed two peaks at similar wavelengths. In this case, the peak at the lower wavelength (331 nm) was approximately double the intensity of the peak at the higher wavelength (494 nm).
The cyclic voltammogram (CV) for the ethylenediamine complex showed that the compound was reduced, but not oxidised on the way back. This has a great deal to do with the reactivity of the central metal ion in different oxidation states.
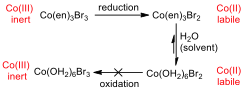
Figure 6 –Oxidation and reduction processes for the Co(en) complex
Cobalt (III) is an inert metal centre, since it is a low spin complex and has six d electrons, it has maximum crystal field splitting energy, and as such, it has little inclination to react in a ligand substitution reaction. When the metal is reduced to Cobalt (II), it becomes much more labile and reactive to substitution, and so all of the ethylenediamine ligands are substituted for water ligands (since water is used as the solvent, it is in great excess). Because the concentration of ethylenediamine is so much lower than that of water, this substitution is essentially irreversible, and the hexaaqua cobalt (II) complex remains in solution.
When the CV tries to oxidise the species back to the original compound, it cannot, because the water ligands cannot be displaced by the ethylenediamine, and the water would be oxidised before the metal centre.
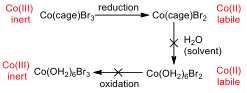
Figure 7 – Oxidation and reduction processes for the cage complexes
Appendix 6 shows the CV for Co(sep)Br3, and in this case, the compound is both reduced and oxidised. This is possible because, even though the metal centre becomes more susceptible to substitution when it is reduced, it cannot escape from the cage ligand, and so substitution for water ligands is not possible. As such, the metal can be re-oxidised, since there are no water ligands that would be oxidized first.
The CV of Co(diNOsar)Br3 shows the same trend as the sepulchrate cage in the range of 0 – -800 mV, however, if the potential is decreased further, it can be shown that the complex can be reduced one more time. This second peak is due to the reduction of the nitro group to the hydroxylamine group. The nitro group is willing to accept an electron because it has many electronegative atoms in it. This reduction is irreversible, since the hydroxylamine group is reluctant to donate an electron (since the electronegative atoms are still present).
Conclusion
The aim of this experiment was to assemble a cobalt cage complex in situ by using smaller, bidentate ligands, and to compare its spectroscopic data to that of stock solutions of the starting material and the product.
The complex was successfully synthesised, and its UV-Vis data was similar to that of the stock solutions of reagent and product, since the metal, its oxidation state, and ligating atoms are the same in all three compounds.
[1] G L Miessler, D A Tarr, Inorganic Chemistry, second edition, Prentice-Hall Publishing, New Jersey, 1998
[2] M Morris, CHM2102 lecture course, University of Sheffield, 2011
[3] P V Bharatam, K Lammertsma, Theoretical and computational chemistry, 12, 2003, pp 61-89
[4] J M Harrowfield, Journal of Chemical Education, 62, 1985, pp 804-806
[5] S Ralph, CHEM301 lecture course, University of Wollongong, 2011
[6] J Clayden, ET. Al., Organic Chemistry, Oxford University Press, 2001
[7] http://en.wikipedia.org/wiki/File:D6_Tanabe-Sugano_diagram.png (accessed 26/10/11)

{kind=link}